Restez toujours informé: suivez-nous sur Google Actualités (icone ☆)
La complexité des communautés microbiennes fait qu'il est difficile de caractériser les génomes des espèces qui les composent, ainsi que d'en suivre la dynamique au cours du temps. L'équipe de Romain Koszul du département Génomes et Génétique de l'Institut Pasteur, vient de démontrer que l'analyse des collisions physiques entre molécules d'ADN d'une population microbienne résout plusieurs des limitations techniques actuelles, permettant notamment d'identifier les hôtes bactériens de phages et plasmides de la population. Ces travaux ont été publiés le 17 février 2017 dans la revue Science Advances.
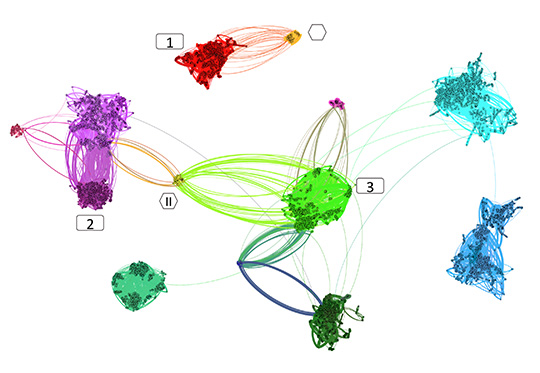
Figure: Avec l'approche de métagénomique dite meta3C (3C pour capture de conformation de chromosome), une carte de contacts entre tous les fragments d'ADN microbiens est établie. Des analyses computationnelles permettent de reconstituer les génomes des différentes bactéries (larges groupes de contigs faisant beaucoup de contacts entre eux) et des phages (qui correspondent a des petits groupes de fragments d'ADN) et d'en déduire quels phages sont en contact avec quelles bactéries... par exemple, l'ADN de la bactérie 1 est en contact avec l'ADN du phage I, ce qui suggère que cette bactérie est l'hôte de ce phage. Les bactéries 2 et 3 sont toutes deux en contact avec l'ADN du phage II, ce qui suggère que ce phage peut infecter ces deux espèces.
© Romain Koszul. Martial Marbouty
Le microbiote intestinal est un élément essentiel de l'écosystème de notre corps. La dynamique, l'équilibre et les effets des communautés microbiennes sont fortement influencés par les phages (les virus bactériens) présents dans la population. C'est pourquoi l'étude des relations bactérie-phage est importante pour comprendre ces écosystèmes dans toute leur complexité, et notamment comment ils évoluent au cours du temps.
La métagénomique est la discipline qui permet l'étude du métagénome, ou l'ensemble des fragments d'ADN issus de populations de microorganismes comme le microbiote. Les limitations dans les techniques de séquençage font qu'il est difficile, d'une part, de caractériser les génomes bactériens et viraux complets à partir d'un mélange complexe d'espèces, et d'autre part, lorsqu'on parvient à déterminer des séquences d'ADN de phages présentes dans une population de bactéries, d'attribuer les séquences de phages à leurs hôtes bactériens. Pour lever ces freins, l'équipe de Romain Koszul a utilisé une nouvelle approche de métagénomique, dite meta3C (3C pour capture de conformation de chromosome), une méthode expérimentale et computationnelle qui exploite les contacts physiques entre les molécules d'ADN pour en déduire la proximité. Lors d'un précédent travail, la technique avait été validée sur des populations bactériennes contrôlées et connues. Dans cette étude, une population microbienne de composition inconnue a été analysée. Le principe repose sur la quantification des contacts entre molécules d'ADN dans la population. Grace à un agent fixatif, les structures 3D de tous les ADN sont gelées, et les fréquences de contacts entre ceux-ci mesurées par séquençage haut debit. Une carte de contacts entre tous les fragments d'ADN de la population est ainsi générée, à partir de laquelle on peut reconstituer, grâce a des algorithmes développés dans le laboratoire, les génomes des différentes bactéries et des phages de la population, et ce en une seule expérience. Puis, en analysant les contacts entre ces génomes, l'équipe est capable d'en déduire quels phages sont en contact avec quelles bactéries...
Une des prouesses de cette étude est de démontrer l'efficacité de la méthode sur un échantillon naturel complexe: le microbiote intestinal de souris. A partir d'une selle de souris, les contacts entre près de 400000 petits fragments d'ADN microbien ont été caractérisés, à partir desquels plus d'une centaine de génomes bactériens et viraux ont été reconstitués. Et les contacts physiques entre ces génomes ont été identifiés.
Ce travail ouvre de nouvelles perspectives pour dresser un tableau complet de la structure génomique de la flore intestinale, avec identification de nouveaux phages et leur spectres d'infection, c'est-à-dire leur capacité a infecter une ou plusieurs bactéries. Ce type d'analyse du microbiote a des implications importantes en santé humaine, en ouvrant la possibilité d'étudier la dynamique de ces écosystèmes. On peut étudier, par exemple, comment des gènes de résistance aux antibiotiques se propagent dans des populations microbiennes complexes dans différentes conditions.
Les travaux de l'équipe de Romain Koszul s'inscrivent dans cette perspective car elle fait désormais partie d'un consortium européen qui rassemble les efforts de recherche en matière de résistance aux antimicrobiens, la Joint Programming Initiative on Antimicrobial Resistance (JPI AMR), et va appliquer cette méthode sur des échantillons humains hospitaliers et d'autres issus de l'agroalimentaire. Les analyses fourniront des informations précieuses sur l'adaptation et l'évolution des espèces présentes dans l'écosystème microbien.
Populaires