Restez toujours informé: suivez-nous sur Google Actualités (icone ☆)
Les neuropathies optiques héréditaires sont des maladies mitochondriales cécitantes, liées à la dégénérescence des nerfs optiques. Des chercheurs de l'Institut MitoVasc et de l'Institut Imagine, identifient avec leurs collaborateurs 3 familles françaises atteintes de cette maladie avec des mutations dans le gène DNM1L. Cette découverte est très surprenante car la fonction de DNM1L est diamétralement opposée à celle d'OPA1, le gène majeur de cette pathologie dont ils avaient caractérisé les premières mutations en 2000. En effet, DNM1L est impliqué dans la fission des mitochondries alors qu'OPA1 intervient dans leur fusion. L'équilibre subtil entre ces deux forces de fusion et de fission est donc un élément clé de la physiologie du nerf optique. Cette étude a été publiée le 23 septembre 2017 dans la revue Brain.
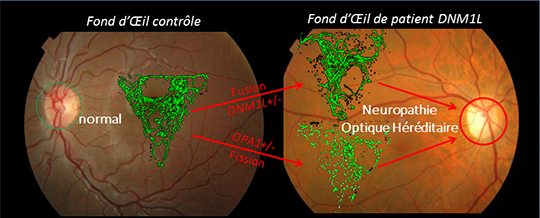
Figure: Réseau mitochondrial (en vert) de fibroblastes sains à gauche et de patients DNM1L (Haut) et OPA1 (Bas) à droite. Notez le teint uniforme de la papille optique chez le patient témoin (cercle vert à gauche) et la pâleur papillaire caractéristique de la maladie chez le patient DNM1L (cercle rouge à droite) reflétant la perte importante des axones des neurones formant le nerf optique. © JM Rozet et G Lenaers.
Les neuropathies optiques héréditaires sont des maladies mitochondriales cécitantes, liées à une dysfonction suivie d'une dégénérescence des nerfs optiques, qui véhiculent l'information visuelle de l'oeil au cerveau. En 2000, les équipes françaises de Christian Hamel et Josseline Kaplan avaient identifié les premières mutations responsables dans OPA1, le gène majeur de cette pathologie retrouvé muté chez 80% des patients, et depuis dans d'autres gènes moins fréquemment mutés.
D'une manière très surprenante, ces mêmes équipes viennent d'identifier 3 familles françaises présentant un tableau clinique identique à celui des patients OPA1, mais avec des mutations dans le gène DNM1L, dont la fonction est diamétralement opposée à celle d'OPA1. En effet, physiologiquement OPA1 est impliqué dans la fusion des mitochondries alors que DNM1L intervient dans leur fission. Par voie de conséquence, les mitochondries apparaissent hyper-fusionnées dans les cellules de patients DNM1L, mais ceci sans conséquence sur l'activité respiratoire de ces mitochondries. L'analyse de souris Dnm1l+/- de 3 mois a confirmé l'allongement des mitochondries dans les nerfs optiques, sans pour autant révéler une dégénérescence significative de ces nerfs.
Donc, l'équilibre subtil entre les forces de fusion et de fission mitochondriale est un élément clé de la physiologie des neurones du nerf optique, probablement pour assurer une distribution mitochondriale et donc énergétique efficiente tout au long des axones de ces neurones, en particulier à l'intérieur de l'oeil où ces axones ne sont pas myélinisés.
Ces découvertes conditionnent d'une manière critique la fenêtre thérapeutique pour traiter les patients atteints de cette maladie.
L'ensemble des signataires de cette publication s'associent pour rendre hommage au Pr. Christian Hamel, décédé cet été, sans qui ce travail et toute la thématique des Neuropathies Optiques Héréditaires n'auraient eu un tel essor scientifique et médical.
Populaires