Unintentional p-value hacking, ou le risque de faire mentir les données, malgré elles
Restez toujours informé: suivez-nous sur Google Actualités (icone ☆)
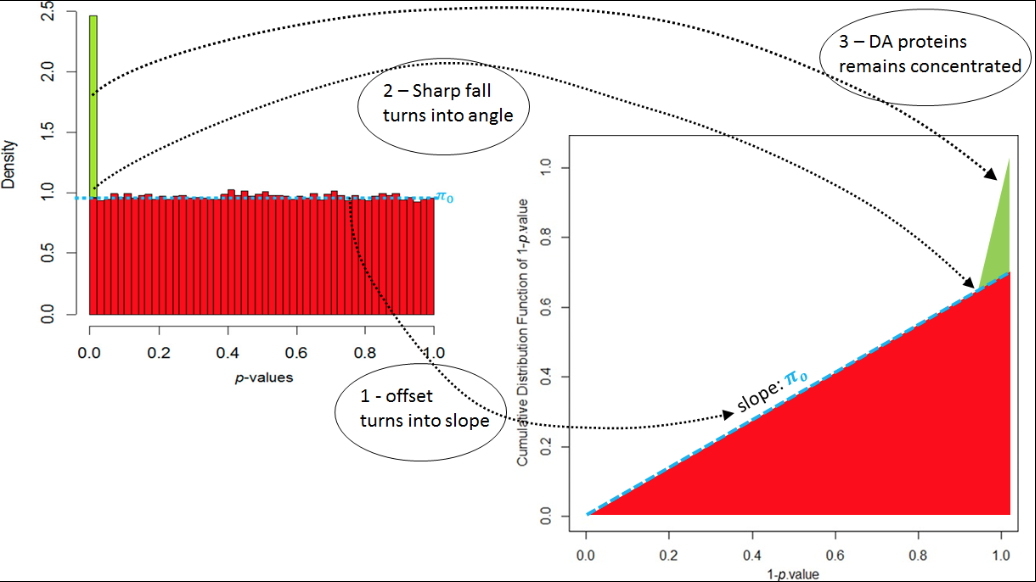
Construction graphique permettant d'estimer visuellement la qualité de la calibration des p-values. © IRIG
"Il y a trois types de mensonges: les mensonges, les sacrés mensonges et les statistiques". Cette phrase de Benjamin Disraeli trouve son origine dans un usage inadapté des outils statistiques, dans le but de cautionner une intuition ou une conviction. Bien que décriée, cette pratique a malheureusement son pendant en recherche: le p-value hacking, qui consiste à triturer les données jusqu'à obtenir la significativité désirée pour une hypothèse testée, au détriment de certaines règles statistiques qui s'en trouvent violées. Le plus souvent, cela est fait en toute bonne foi puisque l'objectif est de tirer le meilleur parti possible des données tout en se conformant aux exigences de publication. Malgré tout, il est indispensable de limiter cette pratique qui engendre de nombreuses fausses découvertes.
Cela est d'autant plus vraie en protéomique, en raison de la complexité intrinsèque du protéome, mais aussi de l'évolution rapide des technologies analytiques. C'est notamment pour cela que de nombreux outils bioinformatiques et biostatistiques fleurissent régulièrement dans la littérature [1], avec la promesse de pouvoir dépasser la triple limite des big proteomics data: leur grande taille, leur grande dimensionnalité, et leur grande complexité. Cependant, la simplicité qu'offrent ses outils ne saurait masquer la nécessité d'un minimum de compréhension théorique pour une utilisation et des résultats corrects.
C'est avec cet objectif que des chercheurs du laboratoire Biologie à Grande Échelle de l'Irig ont réalisé ces dernières années un effort particulier dans la divulgation de bonnes pratiques en science des données pour la protéomique [2-4]. Ils ont ainsi publié une introduction à la théorie du FDR (False Discovery rate, une mesure de contrôle qualité omniprésente), ils ont désambiguïsé un ensemble de termes ayant des significations différentes en intelligence artificielle et en chimie analytique, et ont également proposé cinq étapes de contrôle permettant d'améliorer la qualité d'une analyse protéomique différentielle entre plusieurs échantillons.
Références publication:
[1] Jacob L, Combes F and Burger T. PEPA test: Fast and powerful differential analysis from relative quantitative proteomics data using shared peptides. Biostatistics, 2018
[2] Burger T. Gentle introduction to the statistical foundations of false discovery rate in quantitative proteomics. Journal of Proteome Research, 2018
[3] Borges H, Guibert R, Oermiakova O and Burger T. Distinguishing between spectral clustering and cluster analysis of mass spectra. Journal of Proteome Research, 2019
[4] Wieczorek S, Giai Gianetto Q and Burger T. Five simple yet essential steps to correctly estimate the rate of false differentially abundant proteins in mass spectrometry analyses. Journal of Proteomics, 2019