Valaciclovir - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
| Valaciclovir | |
|---|---|
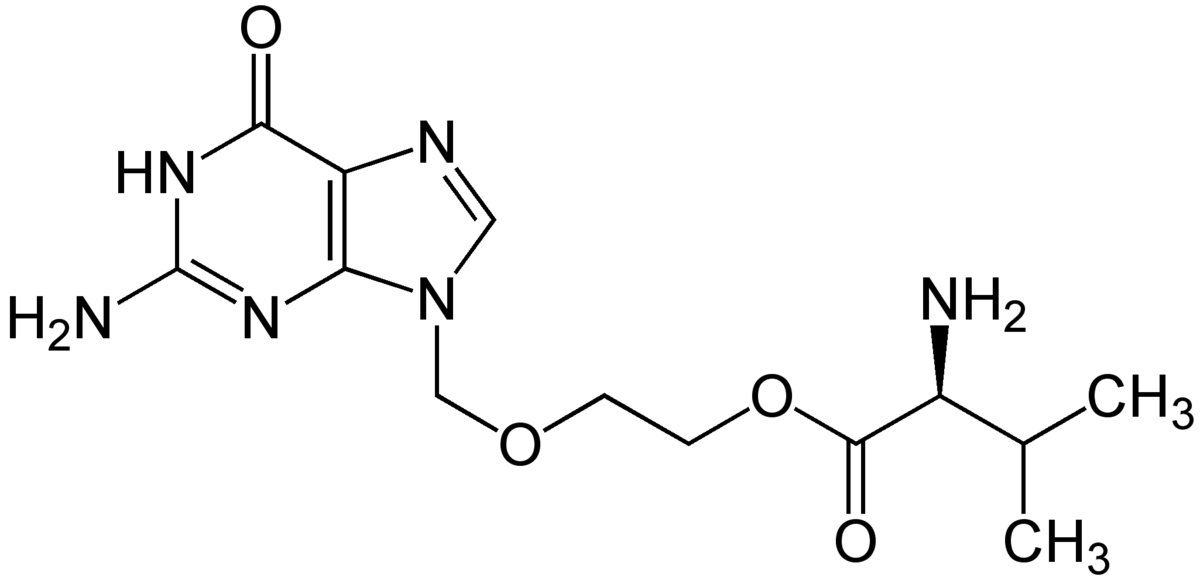
| |
| Général | |
| Nom IUPAC | |
| Synonymes | (S)-2-[(2-amino-6-oxo-6,9-dihydro-3H-purin-9-yl)methoxy]ethyl-2-amino-3-methylbutanoate |
| No CAS | |
| Code ATC | J05 |
| DrugBank | |
| PubChem | |
| SMILES | |
| InChI | |
| Propriétés chimiques | |
| Formule brute | C13H20N6O4 |
| Masse molaire | 324,3357 ± 0,0142 g·mol-1 |
| Classe thérapeutique | |
| Antiviral antiherpétique | |
| Données pharmacocinétiques | |
| Biodisponibilité | 55% |
| Liaison protéique | 13–18% |
| Métabolisme | hépatique (hydrolyse en aciclovir) |
| Demi-vie d’élim. | <30 minutes (valaciclovir) 2,5-3,6 heures (aciclovir) |
| Excrétion | rénal 40–50% (aciclovir) fécal 47% (aciclovir) |
| Considérations thérapeutiques | |
| Voie d’administration | oral |
| | |
Le valaciclovir est un médicament antiviral indiqué dans le traitement du zona, de l’herpès labial (feux sauvages) et de l’herpès génital chez les personnes immunocompétentes, ainsi que pour la suppression de l’herpès génital récurrent chez les personnes infectées par le VIH. Il permet aussi le traitement de certaines infections par cytomégalovirus. C’est un promédicament de l’aciclovir, qui est métabolisé dans le foie en valine et en aciclovir avec une biodisponibilité considérablement supérieure. On note une hydrolyse complète et rapide en aciclovir avant le passage dans la circulation systémique.
Développement
Le valaciclovir a été développé au cours des années 1990 par le laboratoire pharmaceutique Burroughs-Wellcome (devenu GSK) sous la référence BW-256U87. La molécule est un ester formé entre l'aciclovir et l'acide aminé L-valine. Cette combinaison permet d'obtenir une biodisponibilité par voie orale de 50% alors que celle l'aciclovir n'est que de 10 à 20%.
Mécanisme d'action
Le valaciclovir est presque totalement converti en aciclovir lors du premier passage dans le foie. Cet analogue synthétique de la guanine, par hydrolyse, est ensuite phosphorylé par la thimidine-kinase du virus. L’affinité de la molécule pour la thimidine-kinase virale est près de 200 fois supérieure à celle de l’enzyme humaine. La phosphorylation est donc négligeable au niveau cellulaire, réduisant les effets secondaires. Cette affinité sélective est le résultat de l’activation et de la concentration de l’acyclovir dans les cellules infectées. Pour le cytomégalovirus, la phosphorylation est assurée par une activité phospho-transférase dépendantante du gène viral UL97. Après la phosphorylation en aciclo-GMP, les enzymes cellulaires catalysent la phosphorylation en aciclo-GDP et en aciclo-GTP. Le triphosphate d’aciclovir ainsi formé interfère avec l’ADN polymérase de l’herpès virus en tant qu’inhibiteur compétitif sélectif. Il inhibe la réplication de l’ADN viral en compétitionnant avec les désoxyguanosine triphosphates. De plus, l’aciclo-GTP est incorporé dans le brin en élongation et cause la terminaison du brin d’ADN viral. Il réduit ainsi la durée d’élimination du virus et facilite la guérison d’infections primaires cutanées et d’infections cutanéomuqueuses.

















































