Saturation des inégalités d'Heisenberg - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
Le principe d'incertitude d'Heisenberg en mécanique quantique est lié à un théorème d'inégalité. Cette inégalité est dite saturée quand il y a égalité. Quand cette saturation est vérifiée, l'état | ψ > est souvent intéressant à étudier :
Soit A et B deux opérateurs observables qui ne commutent pas, et iC leur commutateur, et soit A et B centrés, alors au mieux
|

Rappel de la démonstration du théorème d'inégalité
L'énoncé du théorème est ( cf principe d'incertitude ) :
![\Delta{A} \cdot \Delta{B} \ge \frac{1}{2} \left| \left\langle \left[ \hat{A}, \hat{B} \ \right] \right\rangle_\gamma \right|](https://static.techno-science.net/illustrationWebp/Definitions/autres/7/7cb2703eac36a28d1371aec39fb4f194_f49520ea2e338fb5dc1c842d7ec34737.png)
Soit A et B deux opérateurs observables qui ne commutent pas : alors on ne peut pas mesurer simultanément A et B ! Le manque de précision est relié à leur commutateur iC. L'opérateur C est hermitien, donc
|

Démonstration :
soit A1 l'observable centrée := A - ; var(A)=
l'inégalité de Schwarz appliquée à |f> := A1| ψ > et |g> = B1| ψ >
donne var(A).var(B) > |
Or A1.B1 = So/2 +iC/2 (avec 2So := A1.B1 + B1.A1, et donc
Donc var(A).var(B) > 1/4
Application à la particule libre
Si la particule est libre, en considérant A= opérateur P et B = opérateur X, on trouve immédiatement :
![[P,X]= -i\hbar](https://static.techno-science.net/illustrationWebp/Definitions/autres/7/7efb94147464594c40e1a0124e6e07c5_8caabcdd0e593494da648741d8075e6a.png)


![\psi(x) = N exp-[\frac{(x-x0)^2}{4v(X)}]](https://static.techno-science.net/illustrationWebp/Definitions/autres/6/6ffb2d80d52b9afca251f77f77658530_41d90fb01d97990a3f8ae15eada223ba.png)
![exp[\frac{ip_0 x}{\hbar}]](https://static.techno-science.net/illustrationWebp/Definitions/autres/b/b656ae8469dc691d5385161f5576bd0f_70f1516446564bb1b5cb6f359e2a2a72.png)
![N = \frac{1}{[2\pi var(X)]^\frac{1}{4}}](https://static.techno-science.net/illustrationWebp/Definitions/autres/b/be7ad1589f069de907d451e14360e70c_31099bd400cf75375dc465d09a1de4b6.png)
C'est le paquet d'ondes gaussien, avec évidemment var(X).var(P)=1/4.

Retrouver une gaussienne n'est pas trop étonnant; mais une faille importante s'introduit dans la dépendance temporelle, à cause de la dispersion inévitable de ψ(x,t) : le paquet d'onde va s'étaler dans le temps. Il faudra donc qu'un potentiel intervienne pour limiter la dispersion : c'est le cas de l'oscillateur harmonique.
Saturation
Il y a saturation si A1 | ψ > = k B1 | ψ > : cet état réalise le minimum d'incertitude.
Par addition , k var(A) +1/k var(B) = 2
d'où la valeur de k : k = i
Alors on dit que A1-k.B1 annihile l'état saturé.
Application à la cuvette 1/n A x^n
A nouveau , l'état fondamental correspondra à la saturation, avec cette caractéristique
-
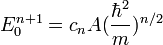
-
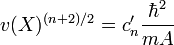
- le paquet d'onde "saturé" reste donc gaussien .


Application à l'oscillateur harmonique
Dans le cas du potentiel 1/2 K x²,on a toujours
var(X).var(P) = 1/4
Donc l'énergie E = 1/2 K.var(X)+1/2 var(P)/m est bornée inférieurement par:

- pour la saturation, E= Eo et l'opérateur d'annihilation donnera encore un paquet d'onde gaussien, mais cette fois la variance est indépendante du temps et on a:
 .
.
- Ce paquet d'onde est stationnaire( l'Énergie est parfaitement connue) et s'étale dans le fond de la cuvette de potentiel; son gradient donne une énergie cinétique moyenne , moitié de l'énergie Eo.
- Remarque: le théorème du viriel, aussi, donne
= = Eo/2. - Ordre de grandeur : dans le cas d'une molécule diatomique covalente, la distance d(A-B) varie toujours un peu à cause de la fluctuation quantique, d'une grandeur dont la variance est, on l'a vu , , où ici m est la masse réduite de A et de B , et K la raideur de la liaison covalente ( typiquement donnée par 2V"(x=d), où V(x) est le potentiel de Morse du modèle d'interaction covalente, soit K = 13.6eV/(0.52Å)^2 = ~ 2000N/m (cf Ordre de grandeur littéral), ce qui conduit à var(X)~ d² 1/1836. A.B/(A+B): inutile donc de donner les distances interatomiques à mieux que 3% .
- Cas de l'helium :Il existe des cas plus surprenants : supposons que la liaison ne soit pas covalente mais plutôt de Lenard-Jones ( modèle des liaisons de Van der Waals) comme dans un cristal de gaz rare: alors K est bien moins élevée et on peut dans le cas de l'hélium se retrouver avec une vibration quantique à température nulle, var(X) ~ d²/10 : or ce critère est celui de Lindemann, pour la possibilité de diffusion des atomes du cristal : celui perd sa cohésion, c'est un LIQUIDE. Ainsi explique-t-on l'anomalie de l'hélium qui est le seul corps impossible à cristalliser par refroidissement seul: il faudra en quelque sorte "raidir" la valeur de K en appliquant une pression pour obtenir l'hélium stable en phase solide.
















































