Détermination des constantes d'équilibre - Définition
La liste des auteurs de cet article est disponible ici.
Mesures expérimentales
La détermination de constantes d'équilibre se fait depuis plusieurs décennies et les techniques utilisées ont beaucoup évolué et sont devenues de véritables spécialités. Ce résumé ne peut pas donner tous les détails nécessaires à l'acquisition de mesures valables, et le lecteur se reportera aux textes spécialisés pour ce faire.
Potentiométrie et pH-métrie
La concentration de certaines espèces peut être mesurée à l'aide d'électrodes spécialisées, telles que l'électrode de verre indicatrice du pH (pour mesurer [H+]) ou les électrodes sélectives (de verre ou à membrane) pour certains autres ions. Ces électrodes devront être calibrées avec des solutions de référence à concentration fixe.
Pour les mesures de pH, des solutions-tampons seront utilisées et les lectures du pH-mètre obéiront
- pH = nF / RT(E0 − E) ,
expression dérivée de l'équation de Nernst, où E0 est le potentiel standard de l'électrode, E est la mesure potentiométrique, n est le nombre d'électrons impliqués (= 1), F est la constante de Faraday, R est la constante de gaz idéal et T représente la température (en kelvin). Chaque unité de pH engendre une différence de potentiel d'environ 59 mV à 298 K. La méthode de Gran peut, à l'aide d'une titration acide fort-base forte, servir de calibration et détecter la présence de carbonates dans un titrant à base d'hydroxyde.
Pour toutes mesures potentio- ou pH-métriques, une calibration simple peut se faire à l'aide de l'équation de Nernst modifiée
- E = a + blog10[A]
où A représente l'espèce détectée par l'électrode. Au minimum deux mesures, depuis au moins deux concentrations de référence, suffisent pour établir les paramètres empiriques a et b permettant ainsi de convertir les potentiels en concentrations.
Le nombre de données doit égaler ou excéder le nombre de constantes d'équilibre à déterminer, ce qui est facile avec une titration.
Limitations
Des valeurs de constantes d'équilibre entre environ 102 et 1011 peuvent être mesurées directement par potentiométrie à l'aide d'électrodes de verre, grâce à la réponse logarithmique de ces électrodes. La méthode par compétition peut étendre cette plage, mais l'équation de Nernst tient mal à très bas ou très haut pH.
Spectrométrie électronique
L'absorbance à une longueur d'onde dans la région visible ou de l'ultraviolet doit obéir la loi de Beer-Lambert, où il existe un rapport linéaire entre la mesure et la concentration de l'espèce mesurée, soit

où


Puisqu'avec une seule mesure, il n'est pas possible de déterminer plus qu'un paramètre, il faut avoir soit pour un même échantillon des mesures d'absorbance à plusieurs longueurs d'onde qui dépendront d'une même valeur de la constante d'équilibre pour ainsi cerner le ou les ε inconnus, soit plusieurs échantillons à différentes concentrations relatives en réactifs dont les mesures d'absorbance dépendront des mêmes valeurs d'ε à la même longueur d'onde pour ainsi cerner la constante d'équilibre inconnue, soit plusieurs échantillons examinés à plusieurs longueurs d'onde. On aura NCNλ mesures d'absorbance provenant de NC solutions (à concentrations relatives différentes) mesurées à Nλ longueurs d'onde, pour évaluer au pire NE(Nλ + 1) inconnus provenant de NE espèces chromophores associées à NENλ valeurs d'ε et NE constantes d'équilibre. Il serait donc souhaitable d'obtenir indépendamment les valeurs d'ε des réactifs isolés (et/ou des produits isolés) et/ou d'évaluer indépendamment, si possible, certaines des constantes d'équilibre, pour ainsi réduire le nombre total d'inconnus par détermination. De toute façon, il faut s'organiser pour avoir autant (ou plus) de données que d'inconnus (au pire NCNλ ≥ NE(Nλ + 1)). On se limite en général à un petit nombre de longueurs d'onde choisies pour bien répondre aux changements des concentrations des espèces chromophores, surtout aux λmax des réactifs purs ou produits isolés.
Limitations
C'est une technique utilisée surtout en chimie de coordination avec les métaux de transition. Une limite supérieure sur K de 104 est habituel, correspondant à la précision des mesures, mais cette limite dépend aussi de l'intensité de l'absorption. Idéalement, les spectres du réactif et du produit sont bien distincts; un chevauchement important nécessite une attention particulière. Soit qu'on obtiendra aussi les spectres individuels du réactif et/ou du produit pour limiter la plage de calcul, et donc les valeurs ε seront connues pour les espèces limitantes, soit le calcul effectuera par là-même une déconvolution du spectre, calcul dangereux à cause de la quantité des ε à déterminer pour obtenir une seule constante d'équilibre.
Spectrométrie par RMN
Cette technique se limite à de cas d'échanges entre espèces relativement simples (par exemple, échanges entre isomères ou entre ligand et complexes), où les noyaux observés changent d'environnement chimique suite à l'échange. À l'idéal, il faut des signaux (préférablement des singulets) bien résolus (sans chevauchement).
S'il s'agit d'un équilibre lent, sur l'échelle de temps du phénomène rmn entre deux espèces détectables simultanément, le rapport de la mesure d'intégration des signaux de chaque espèce suffit à établir le rapport de concentration entre les deux, pourvu que l'on prenne les soins nécessaires à la mesure précise de l'intégration, d'où des mesures à différentes concentrations pourront quantifier la constante d'équilibre entre les deux.
Sinon et habituellement dans les situations de complexation, quand l'équilibre est rapide sur l'échelle RMN, on observe un seul signal par type de noyau pour les espèces en équilibre. Le déplacement chimique de ce genre de signal est alors perçu par l'instrument comme la moyenne

-
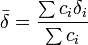

Plusieurs signaux distincts, provenant de noyaux différents dans un même échantillon, peuvent ainsi être soumis au même traitement numérique pour déterminer avec plus de confiance les mêmes constantes d'équilibre. Autrement, plusieurs rapports stœchiométriques seront mesurés.
On aura donc, dans un système d'échange rapide, NCNS mesures de déplacement chimique provenant de NS signaux avec NC échantillons aux concentrations relatives différentes pour évaluer au pire NE(NS + 1) inconnus, soit les déplacements δi des NS signaux des NE espèces en équilibre et NE constantes d'équilibre K. Les valeurs de δ des noyaux dans les espèces limitantes (réactif pur et/ou produits purs) peuvent souvent être déterminées séparément pour limiter la détermination aux valeurs δ et K autrement inconnaissables et donc réduire le nombre de valeurs inconnues, mais de toute façon il faut s'organiser pour avoir autant ou plus de données que d'inconnues (au pire NCNS ≥ NE(NS + 1)).
Limitations
Cette méthode est limitée aux molécules diamagnétiques comprenant un noyau sensible et, ce, soit bien en dessous ou bien au-dessus de la température de coalescence du phénomène d'échange observé. Les déplacements chimiques de référence (par exemple, du réactif isolé, dans une situation de complexation) doivent être obtenus à la même température et dans le même solvant que le mélange contenant un complexe. La précision des mesures de déplacements chimiques convient aux valeurs de K à déterminer allant jusqu'à environ 104, bien que la méthode de compétition peut étendre cette plage.

















































