Méthode du cluster couplé - Définition
La liste des auteurs de cet article est disponible ici.
L'opérateur cluster
L'opérateur cluster est écrit sous la forme :
-
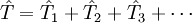

où




et ainsi de suite.
Dans la formule ci-dessus,






Si l'on prend en compte la structure de



Cette série est finie en pratique car le nombre d'orbitales moléculaires occupées est fini, comme d'ailleurs le nombre d'excitations. Afin de simplifier la recherche des coefficients t, le développement de



alors les déterminants de Slater excités plus de n fois peuvent (et habituellement le font) contribuent encore à la fonction d'onde

Description générale de la théorie
La complexité des équations et des codes de calcul correspondants, ainsi que le coût de calcul augmentent rapidement avec le niveau le plus élevé d'excitation. Pour de nombreuses applications, une précision suffisante peut être obtenue par CCSD, et la CCSD(T) plus précise (et plus coûteuse) est parfois appelée « l'étalon-or de la chimie quantique » pour son excellent compromis entre la précision et le coût pour des molécules proches des géométries d'équilibre. Des méthodes plus complexes comme la CCSDT et la CCSDTQ sont utilisées seulement pour des calculs de haute précision sur de petites molécules. L'inclusion de tous les n niveaux d'excitation pour un système à n électrons donne la solution exacte de l'équation de Schrödinger dans la base donnée.
Une amélioration possible pour une approche de cluster couplé standard est l'addition de termes linéaires dans les distances inter-électroniques par des méthodes comme CCSD-R12. Cela améliore le traitement de la corrélation électronique dynamique en satisfaisant la condition de col de Kato et accélère la convergence avec la base d'orbitales. Malheureusement, les méthodes R12 nécessitent la résolution de l'identité qui nécessite une base relativement importante afin d'être valides.
La méthode de cluster couplé décrite ci-dessus est connue comme la méthode de cluster couplé mono-référence (en anglais single-reference (SR) coupled-cluster method car l'ansatz exponentiel n'utilise qu'une fonction de référence