Méthode du cluster couplé - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
| Méthodes numériques pour le calcul de la structure électronique |
| Hartree-Fock |
| Théorie de la perturbation de Møller-Plesset |
| Interaction de configuration |
| Méthode du cluster couplé |
| Champ multi-configurationnel auto-cohérent |
| Théorie de la perturbation multi-référence |
| Théorie de la fonctionnelle de la densité |
La méthode du cluster couplé, ou théorie du cluster couplé (expression souvent abrégée en « cluster couplé », en anglais coupled cluster) est une technique numérique de description des systèmes à plusieurs corps. Son utilisation la plus répandue est comme méthode ab initio de chimie quantique post-Hartree-Fock en chimie numérique. Il est basé sur la méthode d'orbitale moléculaire Hartree-Fock et lui ajoute un terme de correction afin de prendre en compte la corrélation électronique. Certains des calculs les plus précis pour des molécules de petite ou de taille moyenne utilisent cette méthode.
La méthode fut développée initialement par Fritz Coester et Hermann Kümmel dans les années 1950 afin d'étudier les phénomènes de physique nucléaire, mais devint plus fréquemment utilisée après que Jiři Čížek et Josef Paldus eurent reformulé la méthode pour l'adapter à la corrélation électronique dans les atomes et molécules dans les années 1960. Elle constitue à ce jour une des méthodes les plus répandues en chimie quantique incluant la corrélation électronique.
Ansatz de fonction d'onde
La théorie de cluster couplé donne une solution approchée à l'équation de Schrödinger indépendante du temps

où


La fonction d'onde de la théorie du cluster couplé est écrite comme un ansatz exponentiel :
-
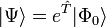

où


Le choix de l'ansatz exponentiel est opportun car (contrairement à d'autres, par exemple, l'interaction de configuration) il garantit l'extensivité de taille de la solution. La consistance de taille, dans la méthode de cluster couplé, dépend de la consistance de taille de la fonction d'onde référence.
Équations de cluster couplé
Les équations de cluster couplé sont des équations dont la solution est un ensemble de coefficients t. Il existe plusieurs façons d'écrire de telles équations mais le formalisme standard conduit à un ensemble final d'équations pouvant être résolu de manière itérative. L'approche variationnelle simple ne tire pas avantage de la nature connectée des amplitudes de clusters et conduit à un ensemble d'équations non achevé.
Supposons qu'il y ait q coefficients t afin de procéder à cette résolution : on a alors besoin de q équations. Il est aisé de noter que chaque coefficient t peut être mis en correspondance avec un certain détermant d'excitation :


où par

Malheureusement,

-
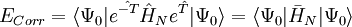
-
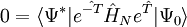


les dernières étant les équations à résoudre et les précédentes l'équation d'évaluation de l'énergie. Considérant la méthode standard CCSD :
-
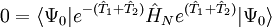
-
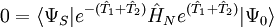
-
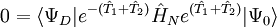



qui lorsqu'elle est factorisée en utilisant la formule de Baker-Campbell-Hausdorff pour le hamiltonien transformé de similarité
![e^{\hat{-T}} \hat{H} e^{\hat{T}} = H + [H,T] + (1/2)[[H,T],T] + ...](https://static.techno-science.net/illustrationWebp/Definitions/autres/f/ffa0ca40683364a985aad7bb96b1b3f1_1269e8fa567556bb5d56a3ce00cc3c77.png)