Réaction d'élimination - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
En chimie organique, une élimination (ou β-élimination) est une réaction qui transforme un alcane substitué (halogénoalcanes, alcools...) en dérivé éthylénique, voire en alcène, si la molécule de départ, outre le groupe partant, n'est qu'une chaîne carbonée de type alcane.
Les conditions sont, en plus dures, celles d'une substitution nucléophile, réaction proche par de nombreux aspects et concurrente : l'élimination se produit en présence d'une base forte et en chauffant le mélange réactionnel.
Équation générale
L'un des principaux exemples d'élimination est la transformation d'un halogénoalcane en alcène ; on parle alors de déshydrohalogénation :
Les bases les plus couramment utilisées sont la soude (Na+,HO-), la potasse (K+,HO-), voire les ions alcoolates (RO-) (obtenus en plongeant Na dans un alcool ROH).
Tout comme pour la substitution nucléophile il existe une élimination monomoléculaire et une élimination bimoléculaire que l'on note couramment E1 et E2 respectivement.
Dans la suite de cet article les exemples sont donnés avec un dérivé halogéné en tant que réactif.
Élimination bimoléculaire
Cinétique
La réaction d'élimination bimoléculaire a une cinétique d'ordre global 2 et d'ordre partiel 1 par rapport au dérivé halogéné R-X et à la base B:-. La réaction se fait en une seul étape qui est bimoléculaire la vitesse de la réaction suit donc une loi de type : v=k.[R-X].[B:-].
où:
- [R-X] représente la concentration en dérivé halogéné en mol.L-1
- [B:-] représente la concentration base en mol.L-1
- v représente la vitesse de la réaction de l'élimination. Elle s'exprime en mol.s-1.
- k est la constante de vitesse. Son unité, L.s-1.mol-1, se déduit de celles des deux autres termes.
Mécanisme de l'E2
L'élimination bimoléculaire est une réaction en une étape, bimoléculaire.

Durant une même étape élémentaire se produisent l'arrachage du proton par la base, la formation de la double liaison, et le départ de l'atome d'halogène (nucléofuge) sous forme d'ion halogénure. On parle de mécanisme synchrone ou concerté.
Stéréochimie
L'E2 est une réaction stéréospécifique.
La réaction ne se fait que lorsqu'un hydrogène et le groupe partant sont en position antipériplanaire.
Facteurs influant sur la réaction
- La vitesse de réaction d'une E2 ne varie que faiblement selon la classe de l'halogénure considéré. Néanmoins, plus la classe est élevée et plus la réaction est rapide.
- La vitesse de réaction croît avec la force de la base.
- Le caractère nucléofuge du groupe partant a également une influence sur la réaction.
Concurrence
Cette réaction est non seulement en concurrence avec l'E1, mais aussi avec les réactions de substitutions nucléophiles, en particulier avec la SN2, qui se produit dans des conditions similaires.
Élimination monomoléculaire
Cinétique de l'E1
La réaction d'élimination monomoléculaire se fait en deux étapes : une étape monomoléculaire qui ne fait intervenir que la molécule du dérivé halogéné du côté des réactifs, qui est l'étape cinétiquement limitante, suivie d'une étape bimoléculaire. Par conséquent la cinétique de la réaction, dans l'approximation de l'étape cinétiquement limitante (ou déterminante), ne dépend que de la concentration de l'espèce RX.
La réaction est donc caractérisée par une cinétique d'ordre global 1, d'ordres partiels 1 par rapport au dérivé halogéné et 0 par rapport à la base B| : v = k.[RX], où:
- [RX] représente la concentration en dérivé halogéné en mol.L-1
- v représente la vitesse de la réaction de l'élimination. Elle s'exprime en mol.s-1.
- k est la constante de vitesse : c'est en fait celle de l'étape cinétiquement limitante (k1). Son unité, L.s-1, se déduit de celles des deux autres termes.
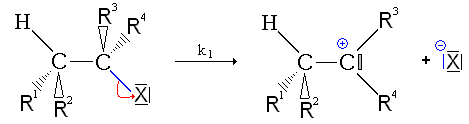
Mécanisme de l'E1
La réaction se fait en deux étapes.
- Première étape : départ du nucléofuge, formation d'un carbocation. cette étape de formation du carbocation est une étape lente, réversible et impose sa vitesse à l'ensemble de la réaction.
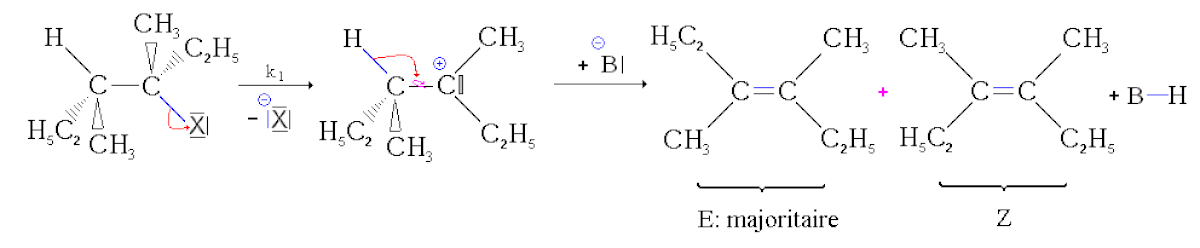
- Deuxième étape : réaction acide-base. La base arrache le proton. Le doublet se "rabat" alors sur la liaison carbone-carbone pour former une liaison double. Comme la liaison simple carbone-carbone peut pivoter, il peut se former deux stéréoisomères. Cette réaction étant beaucoup plus rapide que la précédente, sa vitesse n'influence pas la vitesse globale.
Stéréosélectivité et Régiosélectivité
Stéréochimie
Comme le montre le mécanisme, le passage par un carbocation, qui est une structure plane, enlève tout élément de stéréospécificité contrairement (voir la suite) au mécanisme d'élimination bimoléculaire.
Mais, si deux stéréoiseomères géométriques peuvent se former, ils ne se forment pas dans les mêmes proportions. L'isomère le plus stable, c'est-à-dire celui dont l'énergie est la plus basse, est formé majoritairement. C'est donc l'isomère E qui est majoritairement formé. On parle alors de stéréosélectivité partielle.
Exemple :
Régiochimie
Lorsque plusieurs isomères de constitution peuvent se former, il se forme préférentiellement l'alcène thermodynamiquement le plus stable. S'il n'y a que des groupes alkyles, c'est l'alcène le plus substitué. Si des mésoméries peuvent intervenir, c'est l'alcène le plus conjugué qui est majoritaire. On parle alors de la régiosélectivité ou de la règle de Zaïtsev.
Cependant, parfois l'alcène thermodynamiquement le moins stable se forme préférentiellement, on parle alors de l'élimination d'Hofmann. C'est le cas notamment lorsque la base utilisée est encombrée (ex : KtBuO) ou lorsque que l'on a un ion ammonium qui s'élimine par exemple. (Molécule portant une amine en présence de MeI en nombre d'équivalents suffisant pour former le sel d'ammonium, puis mise en présence d'oxyde d'argent humide et chauffé. Formation de l'alcène par élimination d'Hofmann et donc départ de N(Me)3.)
Facteurs influant sur la réaction
Les paramètres influençant l'élimination E1 sont :
- l'effet de R- : l'étape cinétiquement déterminante est la formation du carbocation (et non sa réactivité), donc plus celui-ci est stable, plus la réaction est favorisée.
- le nucléofuge : la coupure de la liaison C-groupe partant étant l'étape clef du mécanisme, la réaction dépend fortement de la nature de ce nucléofuge. Meilleur est le groupe partant, plus la réaction est rapide. Par exemple pour la déshydratation des alcools, la première étape est la protonation de l'alcool pour former un oxonium, et donc un meilleur groupe partant (H2O>HO-). Un classement rapide des groupe partants serait:
(> meilleur groupe partant que)
À noter que la base n'influence pas ce type de réaction d'ordre 1 et ceci dans la mesure où elle n'intervient pas lors de la première étape qui détermine la vitesse globale de la réaction.
Groupe alkyle du dérivé halogéné
Le mécanisme passe par un intermédiaire réactionnel de type carbocation. Celui-ci est d'autant plus stable qu'il est substitué. Ainsi, la vitesse de réaction (donc de formation du carbocation) progresse des dérivés primaires, aux secondaires puis aux tertiaires. En pratique, seuls ces derniers, et quelques dérivés secondaires, réagissent par ce mécanisme.
Nucléofuge
Plus la liaison R-X est polarisable, plus la rupture est facile. La vitesse croit donc ainsi :

(< : réagit plus rapidement que)
Solvant
- Polarité : un solvant polaire stabilise le carbocation ; il rend ainsi la première réaction plus facile. La vitesse croit donc avec la polarité du solvant.
- Proticité : un solvant protique est plutôt un handicap : en effet il risque de rentrer en concurrence avec le carbocation lors de la réaction acide-base(forte).
Concurrence
Cette réaction est non seulement en concurrence avec l'autre élimination, mais aussi avec les réactions de substitutions nucléophiles, en particulier avec la SN1. Cette dernière se produit en effet dans des conditions similaires (outre le chauffage).

















































