Énergie libre - Définition
En thermodynamique l’énergie libre F (appelée aussi "énergie libre de Helmholtz") est une fonction d’état extensive dont la variation permet d'obtenir le travail utile susceptible d'être fourni par un système thermodynamique fermé, à température constante. Elle correspond à l'énergie libre de Helmholtz des anglo-saxons, qui préférent la symboliser par la lettre A.
À ne pas confondre avec la fonction enthalpie libre G ("énergie libre de Gibbs" des anglo-saxons), qui s’applique aux systèmes évoluant à la température T et à pression constante (cas des réactions effectuées à l’air libre).
L'énergie libre est souvent utilisée dans l'étude des explosions qui induisent une variation de pression ou encore dans la calorimétrie à volume constant effectuée dans une bombe calorimétrique.
Néanmoins le rôle de la fonction F est beaucoup moins important en thermochimie que celui de la fonction enthalpie libre qui est la fonction phare, indispensable à l’étude des équilibres chimiques.
Définition
Considérons une transformation irréversible effectuée à la température T et à volume constant. S’il n’y a pas de montage électrochimique, il n’y a pas de travail électrique. Comme V=cte, le travail des forces de pression est nul.
Donc en appliquant le premier principe :

Appliquons alors le second principe :

Le système échange avec le milieu extérieur Qirrév. Si on se place du côté du milieu extérieur, celui-ci reçoit - Q(irrév) = - ΔU(syst).
Et la variation d’entropie du milieu extérieur devient égale à :

D’où :

Multiplions par ( - T )

On définit ainsi la fonction énergie libre :

|
Pour une transformation effectuée à T et V = cte, on obtient :

Si la transformation est réversible,


En revanche, si la transformation est irréversible,


| La transformation réelle à T et V = cte, ne peut s’effectuer qu’avec une diminution de l’énergie libre du système. |
Différentielle de F

- Appliquons le premier principe

avec:
δWfp : travail des forces de pression
δW': travail autre, comme par exemple le travail électrique dans un montage de pile
- Appliquons le second principe

dU s'exprime donc:

d'où :

|
- Si la température est constante:

- Cas d'une transformation réelle donc irréversible


On montre bien que la variation de la fonction F est égale au travail fourni par le système si la transformation est réversible et est effectuée à T constante .
- Si le volume est constant et le travail W' est nul:

plus précisément :

La transformation réelle à T et V = cte, ne peut s’effectuer qu’avec une diminution de l’énergie libre du système. On peut donc identifier F au potentiel thermodynamique d'une transformation isotherme et isochore
Relations utiles à partir de F ou de ses différentielles
- Relation de maxwell :
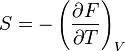

- Relation de Gibbs-Helmholtz : il existe une relation analogue à celle de Gibbs-Helmholtz concernant F :

- Potentiel chimique : une "définition" du potentiel chimique peut être donnée à partir d'une différentielle partielle de F.

Autres fonctions d'état
Réferences
- IUPAC definition
- Atkins' Physical Chemistry, 7th edition, by Peter Atkins and Julio de Paula, Oxford University Press
- Landau, L. D.; Lifshitz, E. M. (1986). Theory of Elasticity (Course of Theoretical Physics Volume 7), (Translated from Russian by J.B. Sykes and W.H. Reid), Third ed., Boston, MA: Butterworth Heinemann. ISBN 0-7506-2633-X.

















































