Fonctionnelle hybride - Définition
La liste des auteurs de cet article est disponible ici.
Fonctionnelles hybrides et connection adiabatique
L'introduction d'un échange HF peut a priori se faire en substituant la partie d'échange des fonctionnelles par l'expression exacte de cet échange :
-
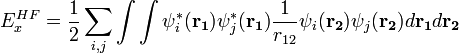

où



Afin de résoudre ce type de problème, A. Becke a mis en avant l'utilité de la formule de connection adiabatique qui s'écrit :

où





-
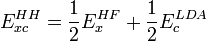

c'est-à-dire l'expression de l'énergie d'échange et corrélation de la fonctionnelle hybride « half-and-half ». Cette dernière a des performances comparables à la GGA aux tests G2, et a de fait ouvert la voie aux fonctionnelles « adiabatiquement connectées » (adiabatically connected functionals en anglais).
Références et notes
- Références et notes citées
- (en) A.D. Becke, « A new mixing of Hartree-Fock and local density-functional theories », dans J. Chem. Phys., vol. 98, 1993, p. 1372–1377
- (en) John P. Perdew, Matthias Ernzerhof et Kieron Burke, « Rationale for mixing exact exchange with density functional approximations », dans J. Chem. Phys., vol. 105, 1996, p. 9982–9985
- (en) Richard M. Martin, Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press, 2004 (réimpr. 2005), 624 p. , p. 165-169
- (en) A.D. Becke, « Density‐functional thermochemistry. III. The role of exact exchange », dans J. Chem. Phys., vol. 98, 1993, p. 5648
- (en) J. Harris et R. O. Jones, « The surface energy of a bounded electron gas », dans Journal of Physics F: Metal Physics, vol. 4, 1974, p. 1170
- (en) O. Gunnarsson et B. I. Lundqvist, « Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism », dans Phys. Rev. B, vol. 13, 1976, p. 4274
- (en) David C. Langreth et John P. Perdew, « Exchange-correlation energy of a metallic surface: Wave-vector analysis », dans Phys. Rev. B, vol. 15, 1977, p. 2884
- (en) J. Harris, « Adiabatic-connection approach to Kohn-Sham theory », dans Phys. Rev. A, vol. 29, 1984, p. 1648
- (en) Oscar N. Ventura et Martina Kieninger, « Computational chemistry as an analytical tool: thermochemical examples in atmospheric chemistry », dans Pure & Appl. Chem, IUPAC, vol. 70, no 12, 1998, p. 2301
- Les tests G2 sont une comparaison des résultats obtenus à la base de données utilisées par J. Pople et collaborateurs pour construire leur méthode Gaussian-2.
- (en) Most Cited Journal Articles 2004-Chemistry and Related, American Chemical Society. Consulté le 10 août 2010
- (en) CAS Science Spotlight Most Cited Chemistry and Related Science, American Chemical Society. Consulté le 10 août 2010
- (en) J. P. Perdew et Y. Wang, « Accurate and simple analytic representation of the electron-gas correlation energy », dans Phys. Rev. B, vol. 45, 1992, p. 13244 .
- (en) A.D. Becke, « Density-functional exchange-energy approximation with correct asymptotic behavior », dans Phys. Rev. A, American Physical Society, vol. 38, no 6, 1988, p. 3098-3100
- Base de données comprenant 56 énergies d'atomisation, 42 potentiels d'ionisation, 8 affinités protoniques et les énergies atomiques des 10 atomes des deux premières rangées du tableau périodique, ayant servi à J. Pople et collaborateurs pour construire leur méthode Gaussian-1.
- (en) P.J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, « Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields », dans J. Phys. Chem., vol. 98, 1994, p. 11623–11627
- (en) Chengteh Lee, Weitao Yang et Robert G. Parr, « Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density », dans Phys. Rev. B, American Physical Society, vol. 37, no 2, 1988, p. 785-789
- (en) S. H. Vosko, L. Wilk et M. Nusair, « Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis », dans Can. J. Phys., vol. 58, 1980, p. 1200
- (en) Markus Reiher, Oliver Salomon, Bernd Artur Hess, « Reparameterization of hybrid functionals based on energy differences of states of different multiplicity », dans Theoretical Chemistry Accounts: Theory, Computation, and Modeling (Theoretica Chimica Acta), Springer Berlin / Heidelberg, vol. 107, no 1, 2001, p. 48-55
- (en) John P. Perdew, Kieron Burke, et Matthias Ernzerhof, « Generalized Gradient Approximation Made Simple », dans Phys. Rev. Lett., American Physical Society, vol. 77, no 18, 1996, p. 3865-3868
- (en) Carlo Adamo, Gustavo E. Scuseria et Vincenzo Barone, « Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model », dans The Journal of Chemical Physics, AIP, vol. 111, no 7, 1999, p. 2889-2899
- (en) "Carlo Adamo, Maurizio Cossi, Giovanni Scalmani et Vincenzo Barone, « Accurate static polarizabilities by density functional theory: assessment of the PBE0 model », dans Chemical Physics Letters, vol. 307, no 3-4, 1999, p. 265 - 271
- Autres références utilisées
- Cécil Combelles, Modélisation ab-initio appliquée à la conception de nouvelles batteries Li-Ion, Université Montpellier II - Sciences et Techniques du Languedoc, 2009, 208 p., p. 35-37.
- Leonard Jeloaica, Etude Ab initio des mécanismes réactionnels dans la phase initiale du dépôt par couches atomiques des oxydes à moyenne et forte permittivité sur silicium, Université Paul Sabatier - Toulouse III, 2006, 141 p., p. 41. thèse de doctorat sous la direction de Mehdi Djafari Rouhani
- Antony Fouqueau, Étude théorique de complexes de Fer par la théorie de la fonctionnelle de la densité: Application au problème de piégeage de spin, Université Joseph-Fourier - Grenoble I, 2005, 264 p., p. 65-66. thèse de doctorat sous la direction de Mark E. Casida
- Fayçal Allouti, Chemins réactionnels conduisant à la formation des oxydes des métaux de transition 3dn (n>5) : structure électronique des composés antiferromagnétiques M2O2 (M=Cr, Mn, Fe, Co, Ni, Cu), Université Pierre et Marie Curie - Paris VI, 2006, 214 p., p. 48-50. thèse de doctorat sous la direction de Mohammad Esmaïl Alikhani

















































