Théorie de l'état de transition - Définition
La liste des auteurs de cet article est disponible ici.
Application de la TST : cas des réactions enzymatiques
Les enzymes catalysent des réactions chimiques avec des cinétiques stupéfiantes comparées à la chimie non catalysée dans les mêmes conditions réactionnelles. Chaque processus catalytique requiert un minimum de trois étapes, toutes se déroulant dans les quelques millisecondes qui caractérisent les réactions enzymatiques typiques. Selon la théorie de l'état de transition, la plus petite partie du cycle catalytique correspond à l'étape la plus importante, celle de l'état de transition. Les propositions initiales de théorie de la constante cinétique absolue pour des réactions chimiques définissaient l'état de transition comme une espèce distincte dans le déroulement de la réaction, espèce déterminant la constante cinétique absolue. Peu de temps après, Linus Pauling proposait que la puissante action catalytique des enzymes puisse être expliquée par une liaison forte et spécifique aux espèces d'état de transition. La constante cinétique étant proportionnelle à la proportion de réactif dans le complexe d'état de transition, l'enzyme était considérée comme pouvant accroître la concentration d'espèces réactives.
Cette hypothèse a été formalisée par Wolfenden et collaborateurs au sein de l'Université de Caroline du Nord à Chapel Hill, qui supposèrent que l'accroissement cinétique imposé par les enzymes est proportionnel à l'affinité de l'enzyme pour la structure de l'état de transition par rapport au complexe de Michaelis-Menten. Les enzymes accélérant les réactions par des facteurs de 1010-1015 par rapport aux réactions non catalysés, et les complexes de Michaelis-Menten ayant parfois des constantes de dissociation de l'ordre de 10−3-10−6 M, il fut proposé que les complexes d'état de transition sont liés avec des constantes de dissociation de 10-14 -10−23 M. Lorsque le substrat évolue du complexe de Michaelis-Menten au produit, la chimie se produit par des modifications enzymatiques dans la distribution électronique dans le substrat.
Les enzymes modifient la structure électronique par protonation, déprotonation, transfert électronique, distorsion géométrique, partition hydrophobe et interaction entre acides et bases de Lewis. Ces actions sont effectuées par des modifications séquentielles des charges protéiques et de substrat. Lorsqu'une combinaison de forces faibles individuelles s'exercent sur le substrat, la somme des énergies individuelles donne des forces capables de relocaliser les électrons liants afin de créer ou de défaire des liaisons. Des analogues ressemblant aux structures d'état de transition pourraient fournir les plus puissants inhibiteurs non-covalents connus, même si seule une petite fraction de l'énergie de l'état de transition est captée.
Toutes les transformations chimiques se produisent via une structure instable appelée état de transition, qui se situe en équilibre les structures chimiques des substrats et des produits. Les états de transitions pour des réactions chimiques sont supposés avoir des temps de vie proches de 10-13 secondes, de l'ordre de durée d'une seule vibration de liaison. Aucune méthode physique ou spectroscopique n'est disponible pour observer directement la structure d'un état de transition pour des réactions enzymatiques, alors que la structure de l'état de transition soit essentiel pour comprendre la catalyse enzymatique, les enzymes fonctionnant en minimisant l'énergie d'activation d'une transformation chimique.
Il est maintenant admis que les enzymes permettent la stabilisation des états de transition se trouvant entre les réactifs et les produits, et qu'elles devraient se lier fortement à tout inhibiteur ressemblant fortement à un tel état de transition. Les substrats et les produits participent souvent à de nombreuses réactions enzymatiques, alors que l'état de transition tends à être caractéristique d'une réaction enzymatique particulière, ce qui rend un inhibiteur spécifique d'une enzyme particulière. L'identification des nombreux inhibiteurs d'état de transition est un argument en faveur de l'hypothèse de stabilisation pour la catalyse enzymatique.
Il y a actuellement un grand nombre d'enzymes connues pour interagir avec des analogues de l'état de transition, la plupart étant conçues pour inhiber l'enzyme cible. On peut citer comme exemple la protéase de HIV-1, les racémases, les lactamases β, les métalloprotéinases, ou encore les cyclooxygénases.
- Cas de la purine nucléoside phosphorylase
La purine nucléoside phosphorylase (PNP) est une enzyme impliquée dans le catabolisme et le recyclage des nucléosides, et constitue une cible pour le développement de nouveaux agents thérapeutiques pour l'apoptose des lymphocytes T dans le cas de la leucémie et les maladies auto-immunes L'inosine, la guanosine et la 2'-déoxyganosine sont les principaux substrats de cette enzyme (la figure 3 montre une réaction catalysée caractéristique de la PNP avec un substrat inosine).
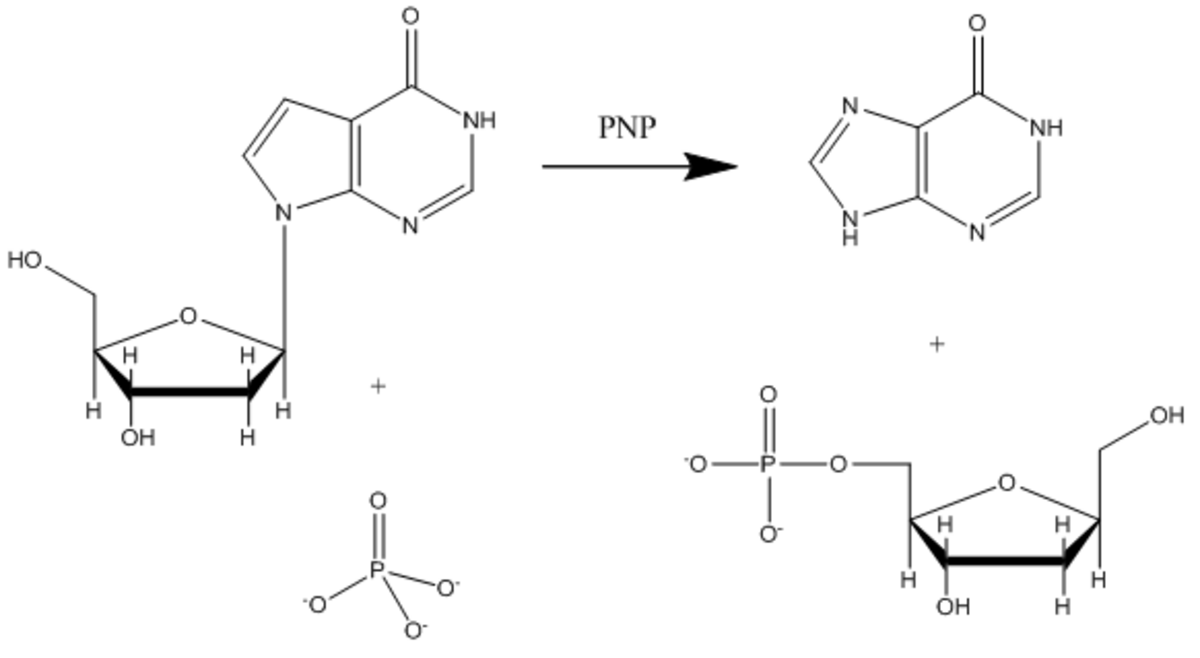
Vern Schramm et ses collaborateurs au sein de l'Albert Einstein College of Medicine ont déterminé la structure de l'état de transition de la PNP et l'ont utilisé afin de développer des analogues à liaisons fortes de l'état de transition afin d'inhiber cette enzyme. L'inhibiteur immucilline H (forodésine) de la PNP est très proche structurellement de l'état de transition putatif (figure 4), avec des modifications nombreuses afin de rendre le composé plus stable que l'éphémère état de transition.
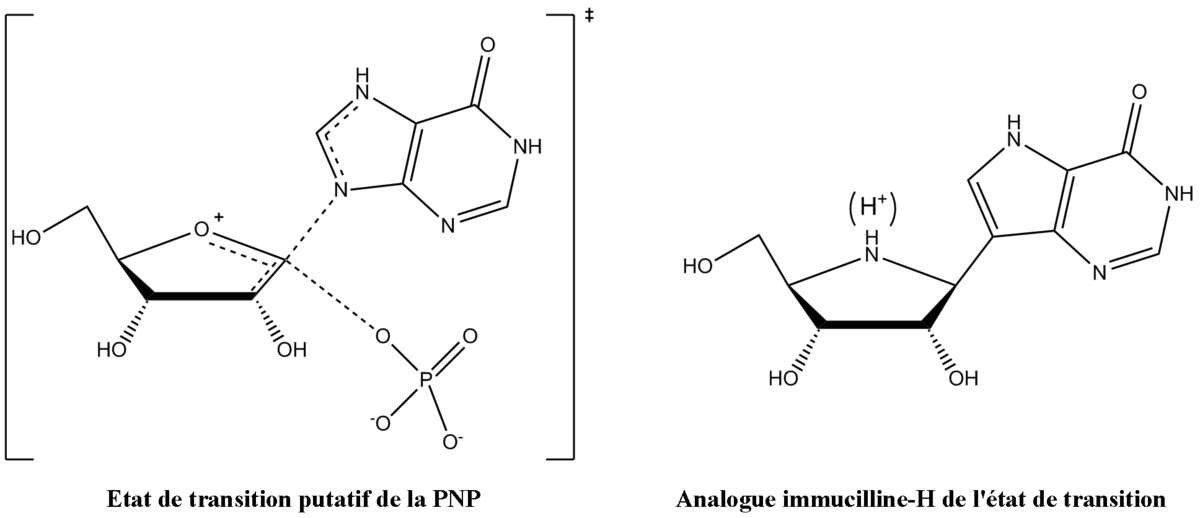
La structure de l'état de transition de la phosphorolyse, stabilisée par la PNP, montre, pour la protonation, un pKa élevé pour la position N7 du cycle purine et sert comme donneur de liaison hydrogène avec l'oxygène de la chaine carbonyle latéral de l'asparagine 243, ce qui est imité dans l'immucilline H par utilisation de la 9-déazapurine en lieu et place de l'hypoxanthine. L'état de transition forme également un ion oxocarbénium dans le cycle de l'ose, qui est fourni par la partie iminoribitol qui possède une liaison ribosidique plus stable. L'ion phosphure n'est pas fortement impliqué dans la formation de liaison dans l'état de transition, et donc a été écarté de la conception de l'analogue de l'état de transition.
Les analogues de l'état de transition ont des propriétés d'inhibiteurs à liaison lente où dans la première étape l'inhibiteur se lie afin de former un complexe réversible EI, qui est suivit par une modification conformationnelle lente qui conduit à un complexe EI* très fortement lié.
-
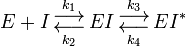
-
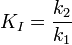
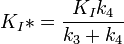



Le KI a été déterminé déterminé par titrage de l'immucilline H et par mesure de son effet sur les vitesses initiales de la PNP vo, et sa valeur est de 41 nM. KI* a été calculé à partir des mêmes mesures cinétiques, mais au lieu d'utiliser les vitesses initiales, les vitesses vs de deuxième étape ont été utilisées, ce qui correspond aux cinétiques inhibées de l'état stable atteint après l'équilibre pour l'étape à initialisation lente lorsque tous les E ont formé des EI.
La stœchiométrie de la liaison de l'immucilline H indiquait qu'une molécule d'inhibiteur se lie à chaque trière de PNP, et une molécule était suffisante pour une inhibition enzymatique. Il avait été démontré auparavant que toute l'activité catalytique de la PNP est supportée par un seul site à la fois, de manière similaire à la protéine F1-ATP synthase. Il a été aussi relevé que la liaison d'analogues du substrat, du produit ou de l'état fondamental peut être réalisée sur les trois sites. Une preuve structurelle appuie l'hypothèse inhibitrice sur un tiers des sites pour cet analogue à l'état de transition, et tous les analogues de l'état fondamental montrent une occupation enzymatique totale.
La conception de l'immucilline H à partir d'une analyse de l'état de transition enzymatique illustre une puissante approche de développement d'inhibiteurs d'enzymes d'intérêt majeur pour l'industrie pharmacologique.

















































