Hypertension artérielle pulmonaire - Définition
La liste des auteurs de cet article est disponible ici.
Physiopathologie
Mécanismes
La pression de l'artère pulmonaire est une pression pulsatile. On définit ainsi, comme pour la pression artérielle systémique (celle que l'on prend classiquement au bras avec un tensiomètre), une pression systolique (la plus grande) et une pression diastolique (la plus petite). On définit également une pression moyenne dont la valeur est intermédiaire des deux autres (la courbe de pression n'étant pas régulière, la pression moyenne n'est pas égale à la moyenne des pressions systoliques et diastolique).
L'hypertension artérielle pulmonaire est définie théoriquement par une élévation de la pression artérielle pulmonaire (PAP) moyenne, qui devient supérieure à 25 mm de mercure au repos ou 30 mm de mercure à l'effort, alors que chez le sujet sain, elle est comprise entre 10 et 15 mm de mercure. Il existe une augmentation de la PAP lors du vieillissement, mais cette augmentation est discrète (de l'ordre de 1 mm de mercure par tranches de 10 années).
La pression artérielle pulmonaire est dépendante de la pression capillaire pulmonaire, du débit sanguin pulmonaire et des résistances vasculaires pulmonaires selon la formule suivante :

avec :
- PAP : Pression artérielle pulmonaire moyenne,
- PCP : Pression capillaire pulmonaire,
-
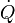
- RVP : résistance vasculaire pulmonaire.

D'après cette formule on déduit le calcul:
- Résistances veineuses pulmonaires (RVP) = (PAPm (mm Hg)-PCAPm (mm Hg)x 80 )/ DC (L/mn)
[normales 90 à 120 dynes-sec-cm-5]. On parle d'hypertension artérielle pré-capillaire pulmonaire si la résistance veineuse pulmonaire est supérieure à 300 dynes-sec-cm-5. Cette dernière peut être indexée avec le débit cardiaque et le poids et la taille du sujet (Index cardiaque) (PAPm (mm Hg) / IC (L/mn/m2) avec une valeur normale comprise entre 2 et 4 Woods). Si elle est supérieure à 10 Woods, il existe une hypertension artérielle pulmonaire. Si les résistances pulmonaires sont basses, l'hypertension pulmonaire est essentiellement en rapport avec une augmentation de la pression du capillaire pulmonaire, témoin indirect de la pression de remplissage du ventricule gauche. Dans ce cas, cette hypertension artérielle pulmonaire est secondaire à une insuffisance cardiaque gauche, a les mêmes causes et répond aux mêmes traitements que pour cette dernière. Le reste de l'article parle donc essentiellement de l'hypertension artérielle pulmonaire de type « pré-capillaire », même si ce n'est pas tout le temps précisé.
La pression artérielle pulmonaire va donc être augmentée en cas d'augmentation :
- de la pression capillaire pulmonaire, secondaire à une augmentation de pression veineuse pulmonaire, elle-même secondaire à une insuffisance cardiaque gauche. On parle alors d'hypertension post-capillaire.
- du débit sanguin pulmonaire, dans certaines cardiopathies congénitales avec une communication entre les cavités cardiaques gauches et droites (communication inter-auriculaire ou inter-ventriculaire, persistance du canal artériel).
- des résistances vasculaires pulmonaires, principalement dans les affections pulmonaires chroniques. On parle d'hypertension pré-capillaire.
Les mécanismes physiopathologiques principaux (en dehors de l'HTAP post capillaire) sont une vasoconstriction, un remodelage vasculaire et des phénomènes de thrombose qui vont progressivement entraîner une augmentation des résistances vasculaires pulmonaires.
La vasoconstriction est un composant précoce de l'hypertension artérielle pulmonaire. Elle peut être l'expression d'un fonctionnement anormal des canaux potassiques ou à un dysfonctionnement de l'endothélium vasculaire (induisant la production chronique de vasodilatateurs). Le remodelage vasculaire implique toutes les couches de la paroi vasculaire et se caractérise par des modifications des tissus (visible par Histopathologie).
L'élévation de la pression artérielle a principalement un retentissement sur le ventricule droit. Lorsqu'elle est saine, cette structure a une paroi mince qui se dilate facilement. Mais l'influence de l'hypertension aboutit à une hypertrophie ventriculaire droite, puis à une insuffisance ventriculaire responsable de dyspnée d'effort (essoufflement à l'effort), d'œdèmes de membres inférieurs, d'une augmentation du volume du foie due à sa congestion (hépatomégalie). À long terme, cela peut entraîner une insuffisance cardiaque irréversible, et même la survenue d'un arrêt cardiaque brutal lors d'un effort.
Anatomo-pathologie
Des anomalies cellulaires ont été décrites dans la vascularisation pulmonaire des sujets atteints et pourraient jouer un rôle dans le développement et la progression de la maladie. Ces anomalies incluent des dysfonctionnement de l'endothélium (couche de cellules bordant l'intérieur du vaisseau sanguin) pulmonaire avec une synthèse anormale substances vasodilatatrices (monoxyde d'azote (NO), de thromboxane A2, de prostacycline et d'endothéline (Neurohormone sécrétée par l'endothélium vasculaire : endothéline-1), une altération des canaux potassiques.
De plus, des lésions se situent dans toutes les différentes structures de la paroi des vaisseaux sanguins et incluent une hypertrophie de la media (secondaire à la fois à une hypertrophie et une hyperplasie (augmentation de volume d’un tissu ou d’un organe due à une augmentation du nombre de ses cellules) des fibres musculaires lisses et une augmentation du tissu conjonctif), un épaississement de l'intima et de l'adventice (par production accrue de matrice extracellulaire) associées à des lésions plus complexes.
Causes
Il existe une classification suivant les causes, établie en 1998 et révisée en 2004 où l'hypertension artérielle pulmonaire est séparée en cinq catégories, elles-mêmes divisées en sous-catégories :
- Hypertension artérielle pulmonaire (groupe 1) :
- forme idiopathique (aucune cause retrouvée et absence d'antécédents familiaux),
- forme familiale,
- associée à une connectivite, à une cardiopathie congénitale, une hypertension portale, une infection par le VIH, à l'utilisation de drogues et toxines, autres (désordres thyroïdiens, maladie de Gaucher, hémoglobinopathies, syndromes myéloprolifératifs),
- associée à une pathologie veineuse ou capillaire (maladie veino-occlusive pulmonaire, hémangiome capillaire pulmonaire),
- hypertension pulmonaire persistante du nouveau-né,
- Hypertension veineuse pulmonaire associée à des maladies du cœur gauche (Groupe 2) :
- maladies de l'oreillette ou du ventricule gauche,
- maladies valvulaires du cœur gauche,
- Hypertension pulmonaire associée à une maladie pulmonaire et/ou une hypoxémie (broncho-pneumopathies chroniques obstructives, syndrome d'apnée du sommeil, maladies interstitielles pulmonaires, exposition chronique aux hautes altitudes) : groupe 3
- hypertension pulmonaire due à une maladie thrombo-embolique (par obstruction thrombo-embolique des artères pulmonaires proximales ou distales, ou par une obstruction d'origine non thrombotique (tumeur, parasite, corps étranger)) : groupe 4)
- Divers (groupe 5) : sarcoïdose, histiocytose X, lymphangiomatose, compression des vaisseaux pulmonaires (adénopathies, tumeur, médiastinite fibrosante).
L'insuffisance cardiaque gauche est la cause de près des trois-quarts des hypertensions artérielles pulmonaires.
Les maladies pulmonaires responsables d'un défaut d'oxygénation du sang (hypoxie) sont la seconde cause des hypertensions pulmonaires : la broncho-pneumopathie chronique obstructive, le syndrome de Pickwick.
Les causes d'hypertension artérielle pulmonaire pré-capillaire incluent l'infection par le VIH (ne donnant une HTAP que dans moins d'1% des cas), la sclérodermie (30% de ces derniers ont une hypertension artérielle pulmonaire) et autres maladies auto-immunes, la cirrhose et l'hypertension portale, la drépanocytose, les pathologies thyroïdiennes, certaines cardiopathies congénitales, la sarcoïdose, l'histiocytose X.
Certains comportements peuvent également déclencher cette variation de pression artérielle tel que la prise de certains médicaments anorexigènes, de cocaïne, de méthamphétamine, d'alcool.
Il existe des formes familiales, liées dans près des deux tiers des cas à une mutation sur le gène BMPR2 (Bone morphogenetic protein receptor type II). La pénétrance de cette mutation est à pénétrance variable : seulement 15 à 20% des sujets porteurs de la mutation génétique développeront la maladie. Une autre forme familiale est dans le cadre d'une maladie de Rendu-Osler.
Lorsqu'aucune cause ne peut être identifiée, on parle d'hypertension artérielle pulmonaire primitive ou idiopathique.
L'incidence des HTAP, dite de group 1, est faible (15 à 50 par millions d'habitants et par an)