Chimie numérique - Définition
La liste des auteurs de cet article est disponible ici.
Études des structures moléculaires et cristallines
Une formule moléculaire ou cristalline donnée peut correspondre en réalité de nombreux isomères ou structures. Chacun d'entre eux représente un minimum local de la surface d'énergie (appelée surface d'énergie potentielle) créée à partir de l'énergie totale (énergie électronique, énergie de répulsion entre noyaux, etc.) en tant que fonction des coordonnées de l'ensemble des noyaux. Un minimum local (d'énergie) est un point stationnaire à partir duquel tout déplacement conduit à un accroissement de l'énergie totale du système. Le minimum local le plus bas est appelé minimum global et correspond à l'isomère le plus stable pour la formulation initiale. S'il existe une modification de coordonnées particulière qui conduit à une décroissance de l'énergie dans les deux directions, le point stationnaire correspond à une structure de transition et la coordonnée est la coordonnée de réaction. Le procédé de recherche des points stationnaires est appelé optimisation de géométrie.
La détermination d'une structure par optimisation de géométrie devient routinière lorsque des méthodes de calcul des dérivées premières de l'énergie selon toutes les coordonnées atomiques efficaces sont disponibles. L'évaluation des dérivées secondes liées permet de prédire les fréquences vibrationnelles si un mouvement harmonique est supposé. Cela permet, de manière simplifiée, de caractériser les points stationnaires. Les fréquences sont liées aux valeurs propres de la matrice des dérivées secondes (matrice hessienne). Si les valeurs propres sont toutes positives, alors les fréquences sont toutes réelles et le point stationnaire est un minimum local. Si l'une d'entre elles est négative, c'est-à-dire correspond à une fréquence imaginaire, le point stationnaire correspond à une structure de transition. Si plus d'une d'entre elles est négative, le point stationnaire est plus complexe, et habituellement de peu d'intérêt. Lorsque ce type de point est découvert, il est nécessaire de déplacer la recherche vers d'autres points si l'on recherche les minima locaux et les structures de transition.
L'énergie totale est déterminée par les solutions approchées de l'équation de Schrödinger dépendante du temps, habituellement avec les termes non relativistes inclus, et souvent en utilisant l'approximation de Born-Oppenheimer qui, en se basant sur la plus grande vélocité des électrons comparée à celle des noyaux, permet la séparation (découplage) des mouvements électroniques et nucléaires, et simplifie de fait l'équation de Schrödinger. Ceci permet d'évaluer l'énergie totale comme une somme de l'énergie électronique pour des positions nucléaires fixes et de l'énergie de répulsion des noyaux. Une exception notable est développée dans certaines approches connues sous le nom générique de chimie quantique directe, qui traitent les électrons et les noyaux sur un pied d'égalité. Les méthodes basées sur la fonctionnelle de la densité et les méthodes semi-empiriques peuvent être considérées comme des variantes sur ce thème majeur. Pour des systèmes très étendus, l'énergie totale est déterminée par mécanique moléculaire. Les différentes techniques pour prédire l'énergie totale des structures sont décrites dans les paragraphes suivants.
Les méthodes ab initio
Les codes utilisés en chimie numérique moléculaire sont basés sur de nombreuses et différentes méthodes de chimie quantique qui permettent la résolution de l'équation de Schrödinger associée au hamiltonien moléculaire. Les méthodes qui n'incluent aucun paramètre empirique ou semi-empirique dans leurs équations, c'est-à-dire qui dérivent directement des principes théoriques, sans inclusion de données expérimentales, sont appelées méthodes ab initio. Cela n'implique pas que la solution obtenue est la solution exacte; elles consistent toutes en des approximations à divers degrés des calculs de mécanique quantique. Cela signifie qu'une approximation particulière est définie de manière rigoureuse sur les premiers principes (théorie quantique) puis résolue avec une marge d'erreur qui est connue de manière qualitative à l'avance. Si des méthodes par itérations numériques sont utilisées, le but est d'itérer jusqu'à atteindre la précision machine.
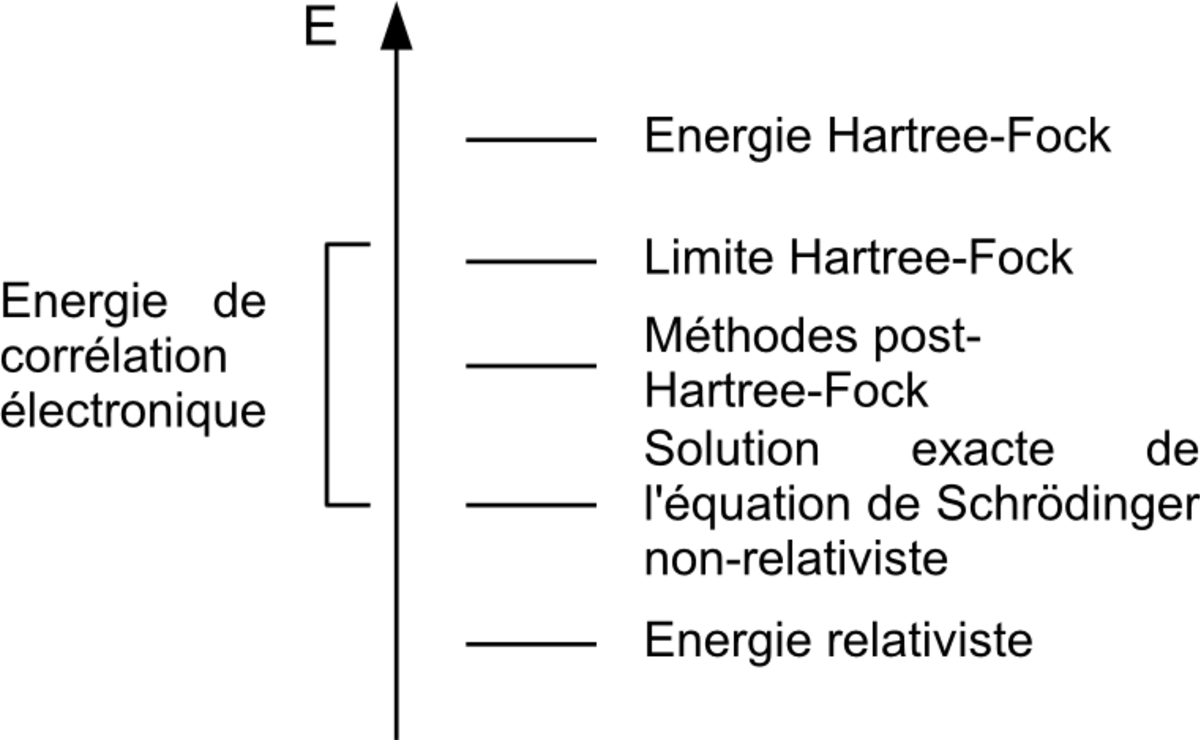
La méthode ab initio la plus simple de calcul de structure électronique est le schéma Hartree-Fock (HF), dans laquelle la répulsion coulombienne électron-électron n'est pas spécifiquement prise en compte. Seul son effet moyen est inclus dans le calcul. Lorsque la taille de la base est augmentée, l'énergie et la fonction d'onde tendent vers une limite appelée limite Hartree-Fock. De nombreux types de calculs, connus sous le nom de méthodes post-Hartree-Fock, commencent par un calcul Hartree-Fock et sont ensuite corrigés pour la répulsion électron-élection, aussi connue comme corrélation électronique. Lorsque ces méthodes sont poussées vers leurs limites, elles approchent de la solution exacte de l'équation de Schrödinger non-relativiste. Si l'on veut obtenir un accord exact avec l'expérience, il est nécessaire d'inclure les termes relativiste et de spin-orbit, les deux n'étant importants uniquement pour les atomes lourds. Dans toutes ces approches, en plus du choix de la méthode, il est nécessaire de choisir une base adéquate. Cette base (au sens mathématique du terme) est un ensemble de fonctions, habituellement centrées sur les différents atomes de la molécule, qui sont utilisées pour étendre les orbitales moléculaires par le postulat de combinaison linéaire d'orbitales atomiques (LCAO). Les méthodes ab initio nécessitent donc d'établir un niveau d'application de la théorie (méthode) et une base.
La fonction d'onde Hartree-Fock est une configuration ou un déterminant simple. Dans certains cas, particulièrement dans les processus de ruptures de liaisons chimiques, elle est relativement inadéquate et de nombreuses configurations sont alors nécessaires. Dans ce cas, les coefficients des configurations et les coefficients des fonctions de base sont optimisés de concert.
L'énergie moléculaire totale peut être évaluée comme une fonction de la géométrie moléculaire, c'est-à-dire, en d'autres termes, la surface d'énergie potentielle. Cette surface peut être considérée dans l'étude de dynamiques de réactions. Dans ce cas, les points stationnaires de cette surface permettent de prédire l'existence de différents isomères ainsi que des structures de transitions pour les conversions entre les différents isomères. Ces propriétés peuvent cependant être déterminées sans la connaissance complète de la totalité de la surface. Dans cette même optique, l'évaluation de quantités thermochimiques dans la limite de précision chimique comme l'enthalpie standard de formation constitue un objectif important. La précision chimique est la précision requise pour effectuer des prédictions théoriques réalistes, et est considérée comme étant de l'ordre de 1 kcal/mol ou 4 kJ/mol. Afin d'atteindre cette précision, il est parfois nécessaire d'utiliser plusieurs méthodes post Hartree-Fock et d'en combiner les résultats. Ces méthodes sont appelées méthodes composites de chimie quantique.
Exemple : Si2H2 ressemble-t-il à C2H2 ?
Des séries d'études ab initio sur Si2H2 (disyline) ont clairement montré la pertinence de la chimie numérique ab initio. Elles s'étendirent sur presque 20 ans, et les conclusions principales furent publiées en 1995. Les méthodes utilisées étaient essentiellement post-Hartree-Fock, en particulier les méthodes d'interactions de configuration (CI) et de clusters couplés (CC). La question initiale était de savoir si Si2H2 possédait la même structure que l'éthyne (acétylène) C2H2. Peu à peu (cette étude ayant commencé avant que l'optimisation de géométrie soit largement répandue), il devint clair que Si2H2 linéaire était en fait une structure de transition entre deux structures trans-déformées équivalentes, et qu'il était en fait moins stable (plus haut en énergie). L'état fondamental fut prédit comme un cycle à quatre déformé en une structure « papillon » avec des atomes d'hydrogènes reliés entre les deux atomes de silicium. L'intérêt dévia ensuite vers le fait de savoir si des structures équivalentes au vinylidène c'est-à-dire Si=SiH2, existent. Cette structure est calculée comme étant un minimum local, soit un isomère de Si2H2, plus élevé en énergie que l'état fondamental mais plus bas en énergie que l'isomère trans-déformé. Puis, de manière surprenante, un nouvel isomère fut prédit par Brenda Colegrove, de l'équipe d'Henry F. Schaefer. Cette découverte fut si étonnante qu'elle nécessita des calculs extensifs pour être confirmée. Il est en effet nécessaire de recourir à des méthodes post-Hartree-Fock afin d'obtenir le minimum local correspondant à cette structure. En effet, ce minimum n'existe tout bonnement pas sur l'hypersurface d'énergie Hartree-Fock. Ce nouvel isomère est une structure plane avec un atome d'hydrogène ponté et un atome d'hydrogène terminal, en position cis par rapport à l'atome ponté. Son énergie est au-dessus de celle de l'état fondamental mais inférieure à celle des autres isomères Des résultats similaires ont été obtenus pour Ge2H2, le germanium appartenant à la même colonne que le silicium dans la classification périodique. De façon peut être plus intéressante, des résultats similaires ont aussi été obtenus pour Al2H2 (puis pour Ga2H2) qui a globalement deux électrons de valence en moins par rapport aux molécules constituées à partir d'éléments du groupe 14. La seule différence est que l'état fondamental du cycle à quatre atomes est plan et non plus déformé. Les isomères cis-mono-pontés et « vinylidène » sont aussi présents. Le travail expérimental sur ces molécules n'est pas aisé, mais la spectroscopie par isolation de matrice des produits de réaction d'atomes d'hydrogène sur des surfaces de silicium et d'aluminium a permis de retrouver les structures cycliques des états fondamentaux ainsi que les structures cis-mono-pontées pour Si2H2 et Al2H2. Les prédictions théoriques sur les fréquences vibrationnelles sont cruciales pour comprendre les observations expérimentales des spectres d'un mélange de composés. Ceci peut apparaître comme un pan obscur de la chimie, mais les différences entre les chimies du carbone et du silicium constituent toujours une question d'actualité, comme d'ailleurs les différences entre les groupes 13 et 14 (principalement entre le bore et le carbone). Les composés de silicium et de germanium ont constitué le sujet d'un article dans J. Chem. Educ..
Méthodes DFT
Les méthodes basées sur la théorie de la fonctionnelle de la densité (DFT) sont souvent considérées comme des méthodes ab initio pour la détermination de la structure électronique moléculaire (ou autre), même si les fonctionnelles les plus courantes utilisent des paramètres dérivés de données empiriques, ou de calculs plus complexes. Ceci permet d'affirmer qu'elles peuvent être aussi qualifiée de méthodes semi-empiriques. Il est sans doute plus pertinent de les considérer comme une classe à part. En DFT, l'énergie totale est exprimée en termes dépendant de la densité électronique plutôt qu'en termes de fonctions d'onde. Dans ce type de calculs, il y a un hamiltonien approximé et une expression de la densité électronique totale également approximée. Les méthodes DFT peuvent être extrêmement précises pour un coût de calcul faible. Le défaut majeur est, contrairement aux méthodes ab initio classiques, il n'existe pas de procédé systématique d'amélioration des méthodes par amélioration de la forme de la fonctionnelle.
Il existe des méthodes (paramétrées) se basant sur une combinaison de la théorie de la fonctionnelle de la densité et de méthode Hartree-Fock pour la description du terme dit « d'échange ». Ces méthodes, développées depuis 1993, sont désignés sous le nom de fonctionnelles hybrides.
Méthodes semi-empiriques et empiriques
Les méthodes semi-empiriques de chimie quantique sont basées sur un formalisme Hartree-Fock, mais procèdent à de nombreuses approximations et utilisent des paramètres issus de données empiriques. Elles sont très importantes en chimie numérique pour traiter de grands ensembles moléculaires dans lesquels une méthode Hartree-Fock pure sans approximations est trop coûteuse. L'utilisation de paramètres empiriques peut permettre d'inclure des effets de corrélation dans les méthodes employées.
Les méthodes semi-empiriques succèdent à ce qui est parfois appelé des méthodes empiriques dans lesquelles la partie à deux électrons du hamiltonien n'est pas incluse de manière explicite. Pour les systèmes à électrons π, il s'agit de la méthode de Hückel proposée par Erich Hückel, et pour tous les systèmes d'électrons de valence, la méthode de Hückel étendue proposée par Roald Hoffmann.
Mécanique moléculaire
Dans de nombreux cas, les systèmes moléculaires importants peuvent être modélisés sans recourir à des calculs de mécanique quantique intégraux. Les simulations de mécanique moléculaire, par exemple, utilisent une simple expression classique pour l'énergie d'un composé, comme celle de l'oscillateur harmonique. Toutes les constantes nécessaires aux calculs doivent être obtenues à partir de données expérimentales ou de calculs ab initio.
La banque de données sur les composés utilisés pour la paramétrisation - l'ensemble de paramètres résultant étant appelé champ de force - est cruciale pour la fiabilité des calculs en mécanique moléculaire. Un champ de force paramétré pour une classe spécifique de molécules, comme par exemple les protéines, ne sera sans doute pertinent que lorsqu'il sera utilisé pour décrire des molécules de la même classe.

















































