Polythiophène - Définition
La liste des auteurs de cet article est disponible ici.
Synthèse
Les PT peuvent être synthétisé par voie électrochimique, en leur appliquant un potentiel sur une solution de monomères afin de provoquer la polymérisation, ou chimiquement, en utilisant des oxydants ou des catalyseurs de couplage croisé. Ces deux méthodes présentent leurs avantages et désavantages.
Synthèse électrochimique
Lors d'une polymérisation électrochimique, un potentiel électrique est appliqué à une solution contenant du thiophène et un électrolyte, produisant ainsi un film PT conducteur à l'anode. La polymérisation électrochimique est intéressante, le polymère n'ayant pas besoin d'être isolé puis purifié, mais elle produit des structures avec des degrés variables d'irrégularités structurales, comme de la réticulation.
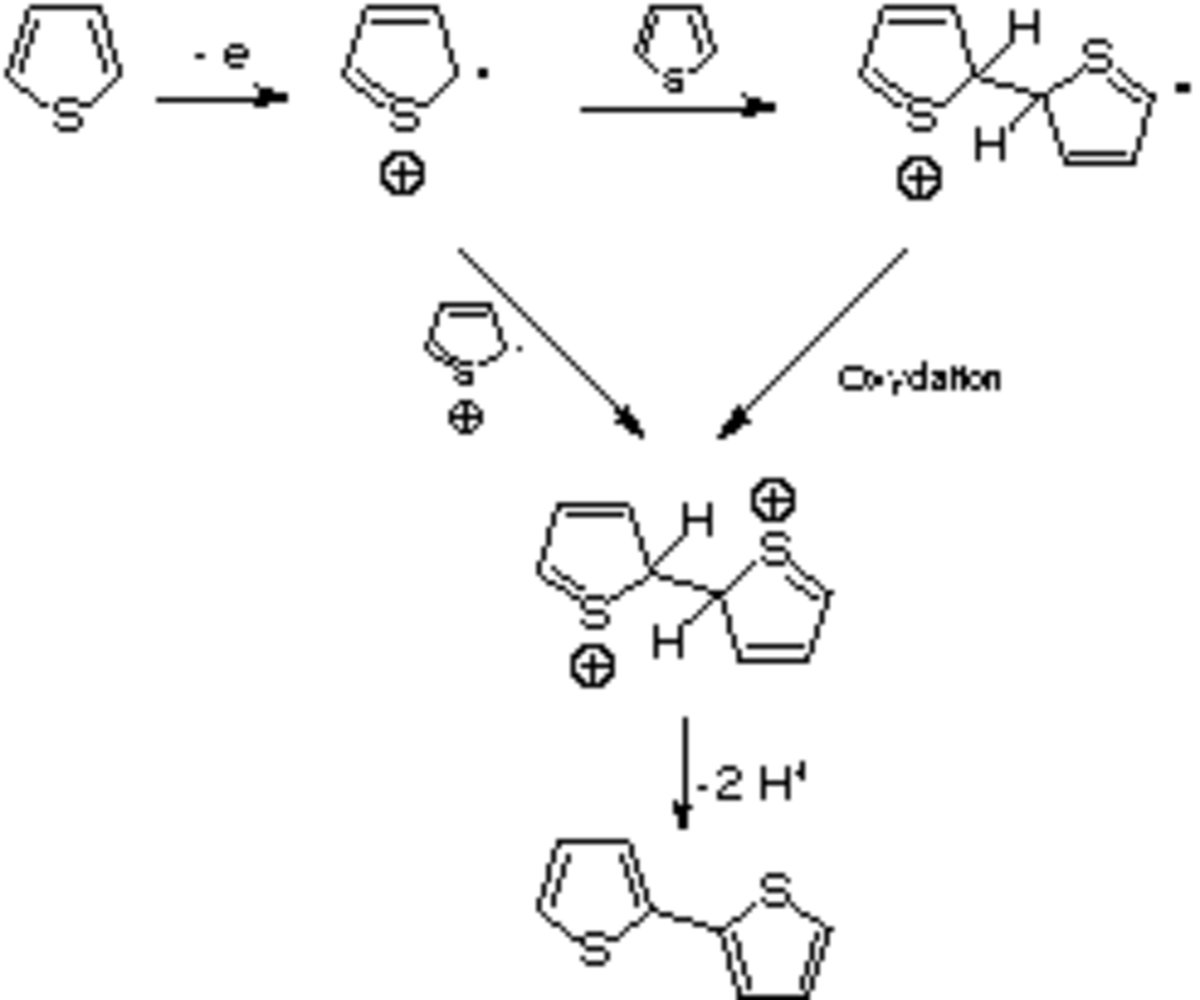
Comme le montre la figure 4, l'oxydation d'un monomère produit un radical cation, qui peut être apparié avec un second radical cation pour former un dimère dication, ou avec un autre monomère pour produire un dimère cationique radicalaire. De nombreuses techniques, comme la microscopie vidéo in situ, la voltampérométrie cyclique, la spectroscopie de photocourant, ou des mesures électrochimiques par microbalance à quartz, ont été utilisées afin de comprendre les mécanismes de nucléation et de croissance conduisant au dépôt du polymère sur l'anode. Le dépôt de longues chaînes bien ordonnées sur la surface de l'électrode est suivie de la croissance de chaînes soit longues et flexibles soit plus courtes et plus réticulées, selon les conditions de polymérisation.
La qualité d'un film de PT préparé par voie électrochimique est dépendante de nombreux facteurs. Cela inclut la matériau d'électrode, la densité, la température, le solvant, l'électrolyte, la présence d'eau et la concentration en monomère. Deux autres facteurs importants mais interagissants sont la structure du monomère et le potentiel appliqué. Le potentiel requis pour l'oxydation du monomère dépend de la densité électronique dans le système p du cycle du thiophène. Les groupes donneurs d'électrons abaissent le potentiel d'oxydation, les groupes accepteurs l'augmentant. En conséquence, le 3-méthylthiophène polymérise dans l'acétonitrile et le tétrafluoroborate de tétrabutylammonium à un potentiel d'environ 1,5 V avec une électrode au calomel saturé (ECS), tandis que le thiophène non substitué polymérise à environ 1,7 V avec une ECS.
La gêne stérique résultant de l'adjonction sur le carbone a d'un thiophène tri-substitué inhibe la polymérisation. Cette observation conduit au phénomène appelé « paradoxe polythiophène » : le potentiel d'oxydation de nombreux monomères de thiophène est plus élevé que le potentiel d'oxydation du polymère résultant. En d'autres termes, le polymère peut être oxydé de manière irréversible et se décompose à un taux comparable à la polymérisation du monomère correspondant. Cela reste un des défauts majeurs de la polymérisation électrochimique, et limite son application pour de nombreux monomères thiophéniques avec des groupes latéraux complexes.
Synthèse chimique
La synthèse chimique des PT présente deux avantages comparé à la voie électrochimique : une meilleure sélection des monomères, et, en utilisant des catalyseurs appropriés, la possibilité de produire des PT substitués parfaitement régioréguliers. Lorsque les PT pourraient avoir été synthétisés chimiquement par accident il y a plus d'un siècle, les premières synthèses planifiées utilisant une polymérisation métal-catalysée du 2,5-dibromothiophène ont été reportées de manière indépendante par deux groupes en 1980. Yamamoto et al. ont utilisé du magnésium dans le tétrahydrofurane (THF) et du nickel-dichlorure de bipyridine, analogue au couplage de Kumada des réactifs de Grignard pour les halogénures d'aryles. Lin et Dudek ont également utilisé du magnésium dans le THF, mais avec une série de catalyseurs acétylacétonate (Pd(acac)2, Ni(acac)2, Co(acac)2, et Fe(acac)3).
Des développements ultérieurs ont produit des PT de plus grande masse molaire que ceux des travaux initiaux, et peuvent être groupés en deux catégories en se basant sur leurs structures. Les PT régioréguliers peuvent être synthétisés par des réactions de couplages croisés catalysés de bromothiophènes, alors que les polymères avec divers degrés de régiosélectivité peuvent être simplement synthétisés par une polymérisation oxydante.

La première synthèse de PAT régioréguliers fut décrite par McCullough et al. in 1992. Comme le montre la figure 5 (en haut), une bromation sélective produit du 2-bromo-3-alkylthiophène, suivie par une transmétallation puis un couplage croisé de Kumada en présence de nickel comme catalyseur. Cette méthode produit environ 100 % de couplages HT–HT, selon l'analyse spectroscopique RMN des diades. Dans la méthode décrite par la suite par Rieke et al. en 1993, du 2,5-dibromo-3-alkylthiophène est traité par le « zinc de Rieke » hautement réactif afin de former un mélange d'isomères organométalliques (figure 5, en bas). L'addition d'une quantité catalytique de Pd(PPh3)4 produit un polymère régioaléatoire, mais un traitement au Ni(dppe)Cl2 produit du PAT régiorégulier avec un rendement quantitatif.
Pour que les méthodes de McCullough et de Rieke produisent des PAT structurellement homogènes, elles nécessitent des températures basses, une déshydratation et une désoxygénation minutieuse, et des monomères bromés. De manière opposée, la polymérisation oxydative des thiophènes utilisant du chlorure ferrique décrite par Sugimoto en 1986 peut être réalisée à température ambiante sous des conditions moins exigeantes. Cette méthode a prouvé son extrême « popularité ». Ainsi, le revêtement antistatique Baytron P de H.C. Stark est préparé à l'échelle industrielle en utilisant du chlorure ferrique (voir ci-dessous).
De nombreuses études ont été menées pour tenter d'augmenter le rendement et la qualité du produit obtenu par technique de polymérisation oxydante. En plus du chlorure ferrique, d'autres agents oxydants, comme l'hydrate de chlorure ferrique, le perchlorate de cuivre et le perchlorate de fer ont été utilisés avec succès afin de polymériser le 2,2’-bithiophène. Une addition lente de chlorure ferrique à la solution de monomère produit des poly(3-(4-octylphényl)thiophène)s avec un taux d'environ 94 % de H–T. La précipitation du chlorure ferrique in situ (afin de maximiser la surface de réaction du catalyseur) donne des rendements et des conversions de monomères significativement supérieurs à l'addition directe du monomère au catalyseur cristallin. Des masses molaires plus importantes ont été obtenues quand de l'air sec a été « bullé » dans le mélange réactionnel lors de la polymérisation. Une extraction de Soxhlet complète après une polymérisation avec des solvants polaires permet de fractionner de manière effective le polymère et d'enlever le catalyseur résiduel avant la spectroscopie RMN. Utiliser un ratio catalyseur/monomère plus faible (2/1, au lieu de 4/1) peut permettre d'accroître la régiorégularité des poly(3-dodécylthiophène)s. Andreani et al. ont indiqué de plus forts rendements pour les poly(dialkylterthiophène)s solubles dans le tétrachlorure de carbone plutôt que dans le chloroforme, qu'ils attribuent à la stabilité des espèces radicalaires dans le tétrachlorure de carbone. Un catalyseur de qualité supérieure, additionné à taux plus faible et à température réduite, a été indiqué comme produisant des PAT de masse molaire plus importante sans résidu polymérique insoluble. Laakso et al. ont utilisé un plan d'expérience pour déterminer que l'accroissement du taux catalyseur/monomère accroît le rendement de poly(3-octylthiophène), et annoncé qu'une durée de polymérisation plus importante augmentait aussi le rendement.
Le mécanisme de polymérisation oxydative utilisant le chlorure ferrique a été controversé. Sugimoto et al. ne spéculèrent aucun mécanisme dans leur publication de 1986. En 1992, Niemi et al. ont proposé un mécanisme radicalaire, indiqué figure 6 (haut).

Ils ont basé leur mécanisme sur deux affirmations. Premièrement, parce qu'ils ont observé la polymérisation uniquement dans des solvants dans lesquels le catalyseur est soit partiellement soit complètement insoluble (chloroforme, toluène, tétrachlorure de carbone, pentane et hexane et pas dans le éther diéthylique, le xylène, l'acétone, ou l'acide formique), ils en ont conclu que les sites actifs de polymérisation devaient être localisés à la surface du chlorure ferrique solide. Donc, ils ont écarté les possibilités de réaction de deux cations radicalaires entre eux, ou de deux radicaux réagissant l'un avec l'autre, « parce que les ions chlorure à la surface du cristal empêcheraient des cations radicalaires ou des radicaux d'occuper des positions compatibles avec la dimérisation ». Deuxièmement, l'utilisation du 3-methylthiophène comme monomère prototype, ils effectuèrent des calculs en mécanique quantique afin de déterminer les énergies et les charges atomiques totales sur les atomes de carbone sur les quatre espèces possibles de polymérisation (3-méthylthophène neutre, cation radicalaire, radical sur le carbone 2 et radical sur le carbone 5).

Puisque le carbone le plus négatif sur le 3-méthylthiophene est le carbone 2, et que le carbone avec la plus forte population impaire d'électrons du cation radicalaire est aussi le carbone 2, ils conclurent que le mécanisme de cation radicalaire conduirait principalement à des liaisons 2–2, H–H. Ils calculèrent par la suite que les énergies totales des espèces avec des radicaux sur les carbones 2 et 5, et trouvèrent que les derniers étaient plus stables de 1,5 kJ/mol. Par conséquent, le radical le plus stable pourrait réagir avec les espèces neutres, formant des couplages tête-queue comme indiqué figure 6 (haut).
Andersson et al. ont proposé un mécanisme alternatif au cours de leurs études sur la polymérisation du 3-(4-octylphényl)thiophène avec du chlorure ferrique, dans laquelle ils obtinrent un haut degré de régiorégularité lorsque le catalyseur était ajouté lentement à la solution de monomères. Ils conclurent que, étant donné la sélectivité des couplages, et les conditions fortes d'oxydation, la réaction pourrait se faire via un mécanisme avec carbocation (voir figure 6, milieu).
Le mécanisme radicalaire fut directement concurrencé dans une communication courte en 1995, lorsque Olinga et François notèrent que le thiophène pouvait être polymérisé par le chlorure ferrique dans l'acétonitrile, un solvant dans lequel le catalyseur est entièrement soluble. Leur analyse de la cinétique de la polymérisation du thiophène semblait aussi contredire les prédictions d'un mécanisme de polymérisation radicalaire. Barbarella et al. étudièrent aussi l'oligomérisation des 3-(alkylsulfanyl)thiophènes, et conclurent d'après leurs calculs de chimie quantique, et à partir des considérations sur la stabilité induite du cation radicalaire lorsqu'il est délocalisé sur un oligomère plan conjugué, qu'un mécanisme par cation radicalaire analogue à celui généralement admis pour la polymérisation électrochimique était plus probable (figure 6, bas). Étant donnée la difficulté d'étudier un système avec un catalyseur hétérogène fortement oxydant qui rend difficile la caractérisation des polymères rigides, le mécanisme de polymérisation oxydante n'est pas encore déterminé. Cependant, le mécanisme par cation radicalaire indiqué dans la figure 6 est communément admis comme la voie de synthèse des PT la plus probable.

















































