Corrosion à haute température - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
La corrosion à haute température est la dégradation des métaux par l'environnement à haute températures (supérieure à 500 °C) ; c'est un phénomène complexe qui a lieu dans les moteurs, chaudières et réacteurs. Les gaz de combustion ont en effet une composition complexe du fait de la composition du combustible et de l'air : N2, O2, CO2 et H2O bien sûr, mais bien souvent également S2, SO2, Cl2, NaCl, et divers oxydes (V2O5...).
On distingue alors deux type de dégradations :
- la corrosion dite « sèche », qui résulte de l'oxydation du métal par les gaz (O2, S2, SO2, H2O) ; on parle aussi d'oxydation à haute température ;
- et la corrosion dite « chaude », ou « fluxage », qui résulte d'une dissolution de l'oxyde par des sels fondus (Na2SO4) et oxydes qui se déposent (il peut aussi y avoir une fusion eutectique, mécanisme similaire au sel sur la glace).
Dans certaines situations, on a cohabitation de métaux solides et de métaux fondus (la température est donc nécessairement élevée). C'est par exemple le cas de la fonderie ; mais les métaux fondus sont parfois utilisés comme fluides, comme par exemple le sodium dans la centrale nucléaire Superphénix. Ces situations entraînent des phénomènes de corrosion particuliers.
La corrosion sèche (oxydation à haute température)
Lorsque l'on met un métal en présence de dioxygène, celui-ci s'adsorbe (c'est-à-dire se fixe) sur la surface et réagit pour former une couche d'oxyde. À température ambiante, la diffusion dans le solide est négligeable ; soit la couche d'oxyde est compacte et protectrice (alumine sur l'aluminium ou chromine sur les aciers inoxydables) et le métal ne bouge pas, soit elle est poreuse ou non adhérente (rouille), et le métal se dégrade par une croissance de la couche d'oxyde au détriment du métal. Les mécanismes qui entrent en jeu sont la migration dans le milieu extérieur (diffusion, convection, champ électrique) et les réactions de surface.
Au-delà de 400 °C, la diffusion en phase solide, qui est activée thermiquement, entre en jeu, et même une couche compacte va pouvoir se dégrader (l'oxyde forme une croûte qui se craquèle).
Pour simplifier, l'étude suivante porte sur l'action du dioxygène seul.
Mécanisme de la dégradation
Dans certains cas, l'oxyde est volatil (cas par exemple du PtO2), ou bien est fragile, poreux, n'adhère pas au substrat. Dans ce cas, le mécanisme de dégradation est évident, le dioxygène réagit avec le métal pour former de l'oxyde et cet oxyde s'évapore ou s'écaille.
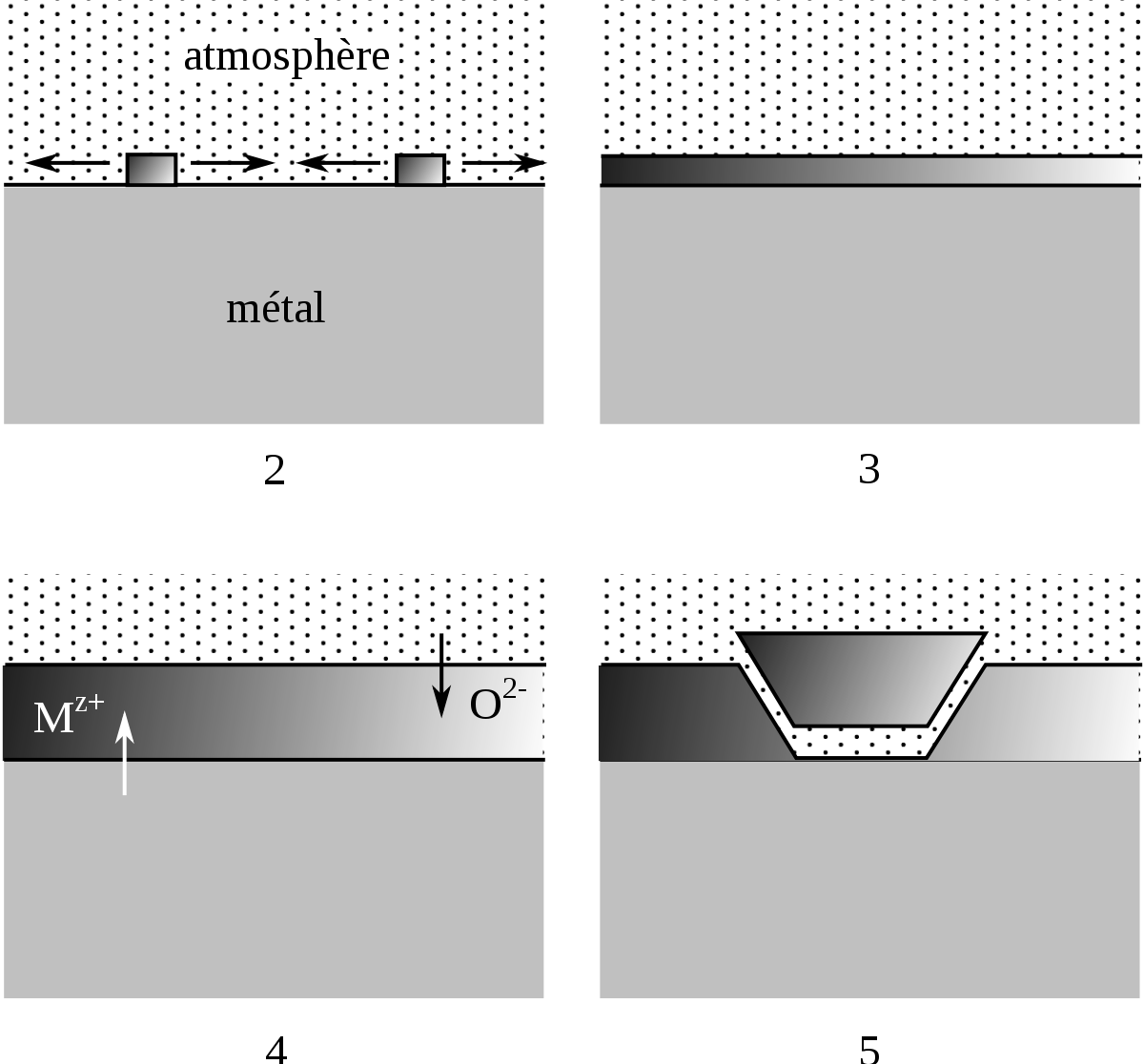
Dans le cas d'un oxyde adhérent et compact, le mécanisme de la dégradation a été décrit par J. Bénard. La dégradation se fait en cinq étapes :
- adsorption et dissociation du dioxygène sur la surface du métal ;
- réaction entre les atomes d'oxygène adsorbés et le métal pour former des germes d'oxyde ;
- croissance latérale des germes jusqu'à la jonction, formation d'un film continu ;
- croissance du film d'oxyde en épaisseur par diffusion dans le film ;
- rupture du film d'oxyde par les contraintes induites par sa croissance et les défauts.

Équilibre thermodynamique : l'oxyde, la forme stable du métal
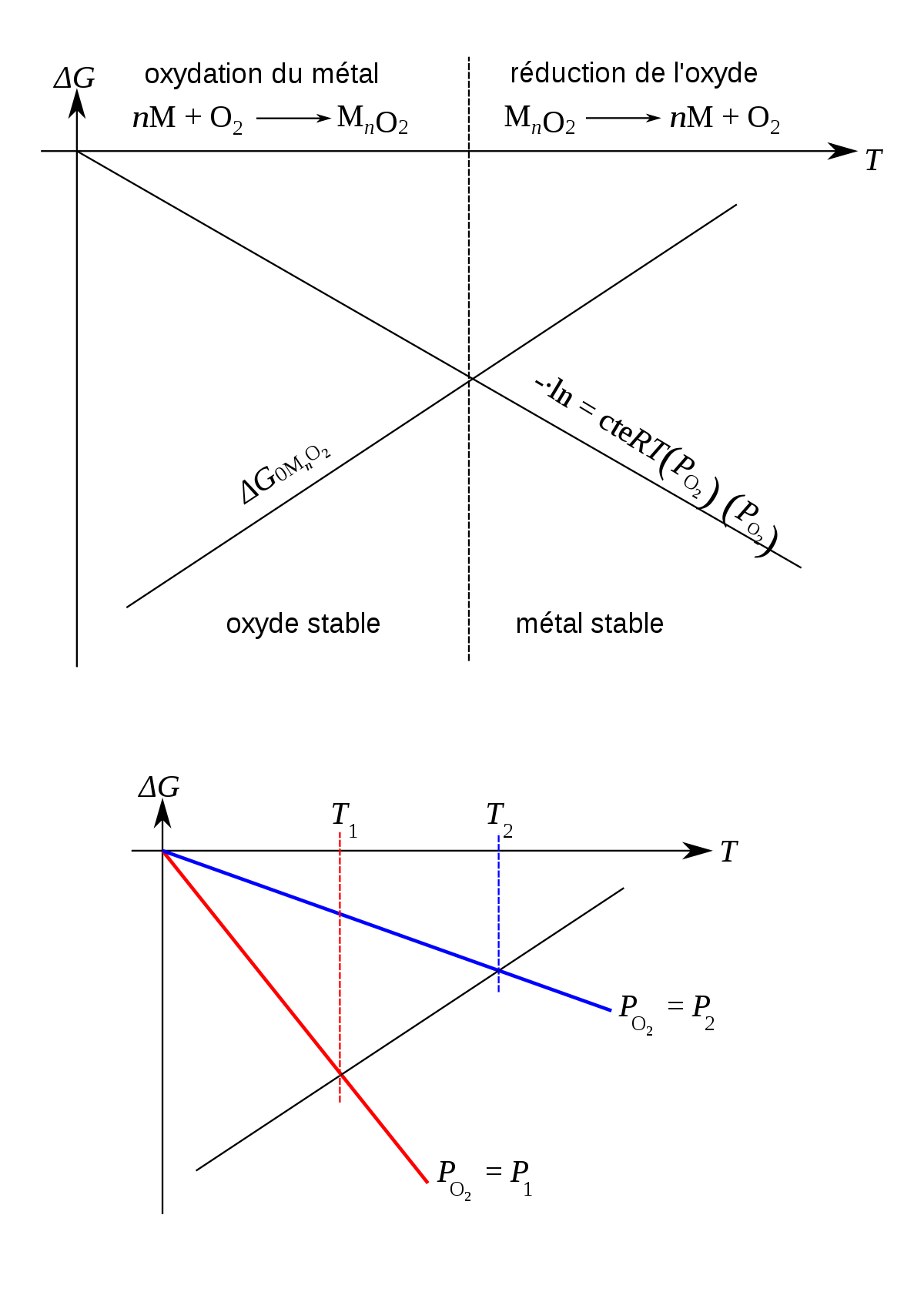
Notons M l'atome de métal, quelle que soit sa nature (Fe, Ni, Al, Cr, Zr…), et notons MnO2 l'oxyde correspondant ; les coefficients ont été choisis pour simplifier l'écriture en considérant la réaction avec une molécule de dioxygène entière, cela peut être Fe2O3, Al2O3, Cr2O3 (n = 4/3), Fe3O4 (n = 3/2), FeO, NiO (n = 2), ZrO2 (n = 1)... L'enthalpie molaire partielle (l'énergie libre de Gibbs) ΔGMnO2 de la réaction d'oxydation
- nM + O2 → MnO2
s'écrit :
- ΔGMnO2 = ΔG0MnO2 + RT·lnPO2
où PO2 est la pression partielle de dioxygène exprimée en atmosphères, R est la constante des gaz parfaits et T est la température absolue exprimée en kelvin (K). L'enthalpie est représentée dans le diagramme d'Ellingham-Richardson, où l'on trace ΔG0(T).
Le diagramme est bâti en supposant l'équilibre thermodynamique, des phases solides pures (activités égales à un), la fugacité du dioxygène égale à sa pression partielle, et que ΔG0 dépend linéairement de la température ; les ΔG se réfèrent à la réaction pour une mole de O2. L'oxydation ne peut avoir lieu que si
- ΔGMnO2 < 0
soit
- ΔG0MnO2 < -RT·lnPO2
Si l'on définit l'enthalpie libre du dioxygène
- ΔGO2 = RT·lnPO2
et que l'on trace -ΔGO2(T) dans ce diagramme, on obtient une droite passant par 0 ; l'intersection de cette droite et de la droite représentant ΔGMnO2 définit la zone de températures où l'oxyde est thermodynamiquement stable pour la pression partielle de dioxygène donnée. Pour les conditions habituelles, la forme stable des métaux est la forme oxydée.
- Note : en toute rigueur, il faudrait noter « -RT·ln(PO2/P0) » où P0 est la pression servant à définir ΔG0 (ou « pression normale »), en l'occurrence 1 atm.
Adsorption du dioxygène et formation des îlots d'oxyde
La molécule de dioxygène O2 se lie au métal puis se dissocie en deux atomes d'oxygène séparés. Les atomes d'oxygène occupent des sites d'adsorption préférentiels, en général les sites ayant le plus grand nombre d'atomes de métal voisins. La répartition de ces sites dépend de la structure cristallographique de la surface, donc notamment de l'orientation du cristallite (ou grain).

Certains auteurs suggèrent que dans le cas d'alliages, les atomes se placent préférentiellement au voisinage des atomes les moins nobles, par exemple le fer dans le cas d'un alliage Fe-Al. Ceci a trois conséquences :
- la structure du germe d'oxyde naissant s'adapte à la structure du substrat, et notamment à l'orientation du cristal métallique (épitaxie) ;
- selon leur orientation, certains germes d'oxyde croissent plus vite que d'autres, le film d'oxyde risque donc de présenter une texture (orientation préférentielle des cristallites) ;
- il peut y avoir un réarrangement des atomes de métal par diffusion de surface (le cristallite change de forme), pour réduire l'énergie d'interface, ce qui peut conduire à la formation de facettes ou de stries.
Une partie de l'oxygène adsorbé se dissout dans le métal et diffuse (c'est-à-dire que les atomes d'oxygène se glissent entre les atomes du métal et progressent vers l'intérieur de la pièce), ce qui dans certains cas peut conduire à une oxydation interne (cf. plus loin).
Croissance latérale des îlots d'oxyde
Les îlots d'oxyde, très minces, croissent latéralement jusqu'à se joindre. Cette croissance se fait par diffusion de surface ; la vitesse de diffusion dépend donc de la densité atomique de la surface. Ainsi, selon l'orientation cristalline du substrat, certains germes d'oxyde croissent plus vite que d'autres. Le film d'oxyde initial peut donc présenter une texture (orientation cristallographique préférentielle).
Croissance du film d'oxyde en épaisseur
Lorsque la couche est adhérente et compacte, l'oxyde isole maintenant le métal de l'atmosphère. Les atomes de dioxygène s'adsorbent donc sur l'oxyde. Lorsque l'oxyde est compact et adhérent, on peut envisager deux mécanismes de croissance :
- l'oxygène adsorbé sur l'oxyde se dissocie, passe en solution dans l'oxyde, diffuse vers l'interface métal/oxyde, et se combine à cette interface avec les atomes de métal ; la création d'oxyde se fait donc à l'interface métal/oxyde, on parle de « croissance vers l'intérieur » ;
- le métal à l'interface métal/oxyde passe en solution dans l'oxyde, diffuse vers l'interface oxyde/gaz, et se combine à cette interface avec l'oxygène adsorbé ; la création d'oxyde se fait donc à l'interface oxyde/gaz, on parle de « croissance vers l'extérieur ».
On peut aussi avoir une combinaison des deux, avec l'oxyde qui se forme au milieu de la couche d'oxyde.
On considère couramment que l'oxyde MnO2 est un composé ionique O2-/Mm+, m respectant la neutralité des charges (m×n = 4) ; la liaison oxyde est en fait plus complexe, mais cette approximation simplifie les calculs de diffusion. La diffusion des espèces se fait donc également sous forme ionique, essentiellement sous forme interstitielle ou lacunaire ; la présence de défauts d'antisites OMm+n' et MOm+n• dans l'oxyde n'est pas envisagée du fait de l'énergie qu'il faudrait pour les créer (on utilise la notation de Kröger et Vink, recommandée par l'Iupac).
Croissance vers l'extérieur
Dans le cas d'une diffusion vers l'extérieur, les ions métalliques partant laissent derrière eux des lacunes. On a donc une contraction de la couche superficielle du métal qui crée des contraintes. Lorsque la concentration en lacune est suffisante, elles se condensent pour former des pores (principe similaire à la précipitation). On constate donc fréquemment des pores à l'interface métal/oxyde. Cette formation de pores provoque une relaxation des contraintes, mais donne lieu à des concentrations de contraintes.
La croissance vers l'extérieur peut se faire de deux manières :
- le gaz réagit avec l'oxyde (le dioxygène est réduit), il capte des atomes métalliques de l'oxyde, laissant des lacunes ainsi que des trous d'électron ;
O2 + MM (oxyde) → 2 O2- + VM (oxyde) + 2h
les lacunes et les trous d'électron diffusent vers l'intérieur, et à l'interface métal/oxyde, des atomes du métal s'oxydent (captent les trous d'électron) pour devenir un ion Mm+ et passe dans l'oxyde, laissant une lacune
M(métal) + m h → Mm•(métal)
Mm•(métal) + VM (oxyde) → MM (oxyde) + VM (métal) ;
en quelque sortes, la réduction du dioxygène crée un déficit d'ion métallique dans la couche, qui « aspire » les atomes du métal ; - un atome du métal passe en insertion dans la couche d'oxyde, diffuse vers l'extérieur et réagit avec le dioxygène en surface de l'oxyde.
Croissance vers l'intérieur
Dans le cas d'une diffusion vers l'intérieur, les ions d'oxygène s'« incrustent » dans le métal et créent donc une dilatation, qui génère des contraintes.
La croissance vers l'extérieur peut se fait de la manière suivante :
- à l'interface métal/oxyde, les atomes du métal réagissent avec les ions O2- de l'oxyde (le métal s'oxyde), laissant des lacunes ainsi que des électron libres ;
nMM (métal) + 2OO (oxyde) → 2 nMM (métal) + 2OO (oxyde) + VO + me'
en quelque sorte, les ions oxygène de l'oxyde OO jouent le rôle de catalyseur ; - les lacunes et les électrons diffusent vers l'extérieur, et à l'interface oxyde/gaz, des molécules du gaz se réduisent (captent les électron) pour devenir des ions O2- et passent dans l'oxyde
O2 + 4e' → 2O2-
O2- + VO (oxyde) → OO (oxyde)
En quelque sortes, l'oxydation du métal crée un déficit d'ion oxyde dans la couche, qui « aspire » les atomes du gaz.
L'autre situation (diffusion d'un atome ou d'un ion d'oxygène en intersticiel) est peu probable, l'oxygène étant un « gros » atome.
Cinétique d'oxydation
Cinétique d'adsorption
L'adsorption du dioxygène peut se décrire par deux phénomènes : d'abord une physisorption : la molécule O2 se lie au métal par une force de van der Waals, de manière réversible, puis une chimisorption, réaction thermiquement activée
- O2 + <
> = < - <
> = 2<> dissociation (chimisorption)
« s » désigne un site d'adsorption, et les doubles crochets <<...>> indiquent que l'espèce est à l'interface métal/gaz. Plusieurs modèles décrivent la cinétique d'adsorption isotherme :
- adsorption monocouche : Hill, Hill et Everett, Langmuir
- adsorption multicouche : BET (Brunauer, Emmet et Teller), théorie de la lame (Frenkel, Hasley et Hill), potentiel Polyani.
mais ils sont rarement utilisés dans ce cadre. En effet, dans notre cas, nous pouvons retenir les hypothèses suivantes :
- la diffusion dans le gaz est rapide et n'est pas un facteur limitant ;
- on est quasiment instantanément à l'équilibre adsorption ↔ désorption, les processus de d'adsorption étant thermiquement activés.
Si le facteur limitant du phénomène est une réaction de surface, on a alors une cinétique linéaire : étant à l'équilibre, l'apport en gaz sur la surface et la quantité de matière se désorbant sont constantes, donc les concentrations en réactants sont constantes. Dès lors, la quantité de matière réagissant est déterminée par la quantité de matière arrivant sur la surface et en partant. Ce flux étant constant (équilibre), on en conclut que la réaction suit une cinétique linéaire :
- mox = kl.t
où mox est la masse d'oxyde, kl le coefficient linéaire d'oxydation et t est le temps.
La cinétique d'adsorption joue dans les cas où l'on a une couche d'oxyde non protectrice (poreuse ou non adhérente, ou bien oxyde volatil) : si la couche est protectrice, la diffusion dans la couche d'oxyde est beaucoup plus lente que l'adsorption et c'est donc la cinétique de diffusion qui contrôle le phénomène. Cependant, la cinétique d'adsorption contrôle les premières minute de l'oxydation, pendant la germination de l'oxyde et la croissance latérale des grains ; certains auteurs ont relevé une cinétique linéaire dans les premières minutes de l'oxydation même dans le cas d'un oxyde compact et adhérent.
Couche adhérente et compacte
La formation initiale du film d'oxyde ne dépend que de l'alimentation en gaz de la surface, et est donc globalement linéaire. Une fois ce film formé, il constitue une barrière entre le métal et le gaz, à condition que ce film soit adhérent et compact. Il y a donc un ralentissement de la corrosion.
Globalement, la corrosion se fait par diffusion à travers l'oxyde. Plus le film est épais, plus le temps de diffusion est long. Une analyse rapide montre que l'épaisseur e du film d'oxyde, et donc la prise de masse de la pièce, varie comme la racine carrée du temps :
- ;
-
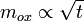

Le premier modèle à cinétique parabolique a été proposé par Tamman en 1920.
En 1933, Carl Wagner a fait une analyse plus fine et obtient lui aussi une cinétique parabolique. Il pose comme hypothèses que :
- la migration fait intervenir, outre la diffusion par sauts aléatoires, l'effet du gradient de potentiel chimique ainsi que l'effet du champ électrique local créé par la répartition des charges ;
- l'oxyde a une composition proche de la stœchiométrie ;
- à tout instant, l'oxyde est localement à l'équilibre chimique ;
- le circuit est ouvert, c’est-à-dire que le courant électrique global est nul et donc que les flux d'espèces chargées sont couplés.
La théorie de Wagner présente l'intérêt de relier la constante de vitesse (constante de proportionnalité entre l'épaisseur et la racine carrée du temps) aux paramètres fondamentaux du matériau (comme les coefficients de diffusion). Dans les faits, cela donne d'assez mauvais résultats, les hypothèses de Wagner étant trop éloignées de la réalité (il ignore en particulier le rôle des joints de grain dans la diffusion).
Mais on constate cependant bien expérimentalement une croissance en racine carrée du temps.
Rupture de la couche ou couche poreuse
Lorsque la couche se rompt, en raison des contraintes générées, le gaz accède directement à une surface importante non-oxydée. On constante donc une accélération de la prise de masse, une rupture de la loi quadratique.
Lorsque la couche est très fragile et se rompt ou se décolle en permanence, ou bien lorsque l'oxyde est poreux, voir volatil, rien ne s'oppose à l'oxydation, la loi est donc linéaire.

















































