Spectroscopie rotationnelle - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
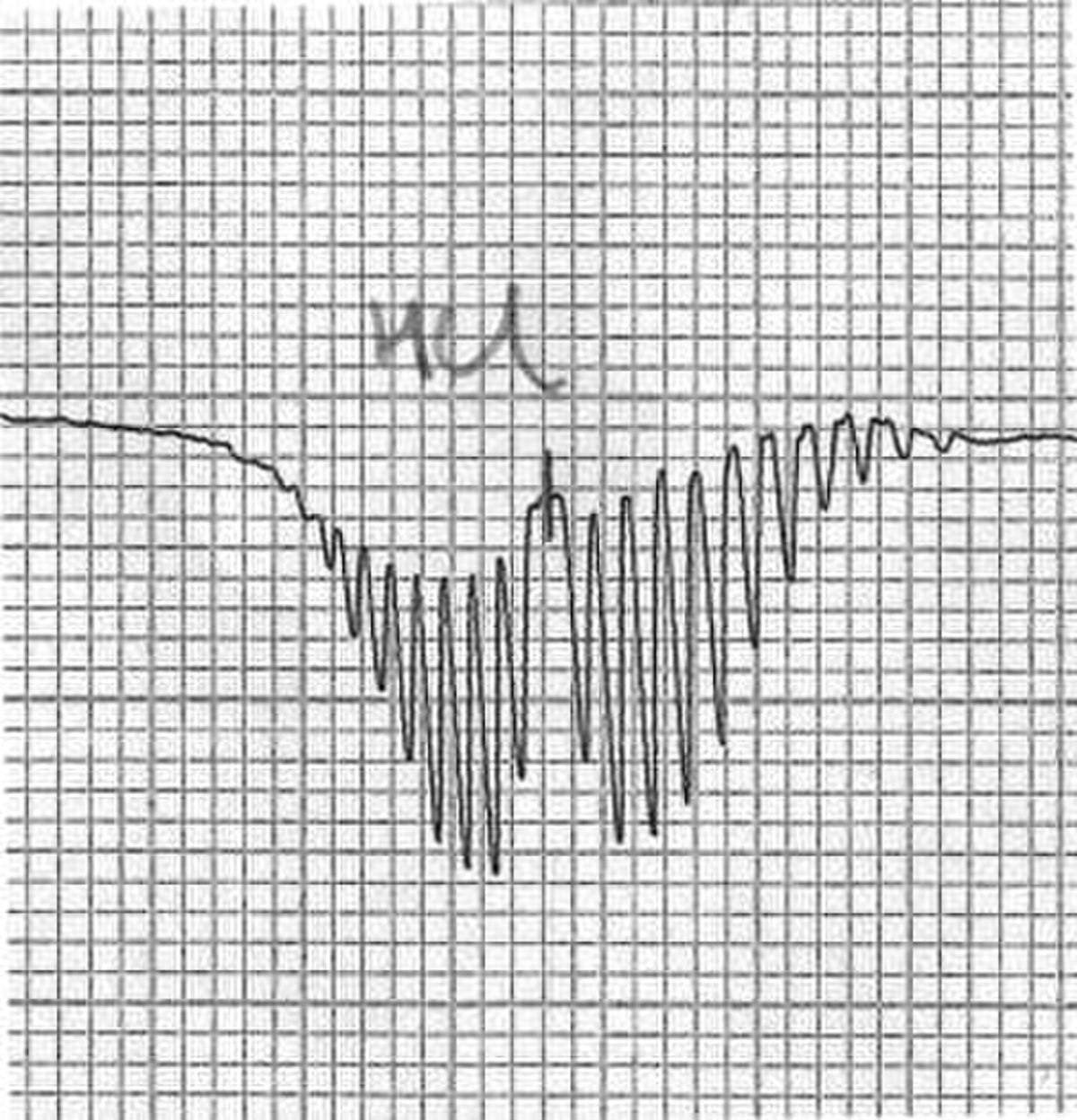
La spectroscopie rotationnelle, de rotation ou micro-onde étudie l'absorption et l'émission d'une onde électromagnétique (habituellement dans la région microonde du spectre électromagnétique) par des molécules associées aux modifications correspondantes du nombre quantique de rotation de la molécule. L'utilisation de micro-ondes en spectroscopie a été rendue possible en raison principalement du développement de la technologie associée pour le RADAR durant la Seconde guerre mondiale. La spectroscopie rotationnelle n'est réellement possible qu'en phase gazeuse quand le mouvement de rotation est quantifié.
Le spectre de rotation d'une molécule (au premier ordre) nécessite que la molécule possède un moment dipolaire et que les centres de charge et masse soient distincts, ou, de manière équivalente, qu'il existe une différentiation de deux charges distinctes. C'est l'existence de ce moment dipolaire qui permet au champ électrique de la lumière (micro-onde) d'exercer un couple sur la molécule, ce qui la fait tourner plus rapidement (en excitation) ou lentement (en désexcitation). Les molécules diatomiques comme le dioxygène (O2) ou le dihydrogène (H2) n'ont pas de moment dipolaire et par conséquent n'ont pas de spectre purement rotationnel. Cependant, les excitations électroniques peuvent conduire à une distribution asymétrique de charge et donc induire un moment dipolaire. Dans de telles circonstances, ces molécules auront un spectre rotationnel.
Les molécules diatomiques hétérogènes comme le monoxyde de carbone (CO) possède l'un des spectres de rotation les plus simples. C'est également le cas pour des molécules triatomiques comme le cyanure d'hydrogène (HC≡N) pour les molécules linéaires ou l'isocyanure d'hydrogène (HN=C:) pour les molécules non-linéaires. Plus le nombre d'atomes croît, plus le spectre devient complexe, les raies dues à différentes transitions commençant à se superposer.
Comprendre un spectre de rotation
En mécanique quantique, la libre rotation d'une molécule est quantifiée, ce qui fait que l'énergie de rotation et le moment angulaire ne peuvent prendre seulement que certaines valeurs fixes. Ces valeurs sont liées de manière simple au moment d'inertie, I, de la molécule. En général pour une molécule quelconque il y a trois moments d'inertie IA, IB et IC liés à trois axes orthogonaux A, B et C avec pour point d'intersection le centre de masse du système. Les molécules linéaires constituent un cas à part. Ces molécules possèdent une symétrie cylindrique et un de ces moments d'inertie (IA, qui est le moment d'inertie pour une rotation autour de l'axe de la molécule) est négligeable (i.e.

Classification des molécules selon leur comportement rotationnel
La convention générale est de définir les axes tels que l'axe A possède le plus faible moment d'inertie (et donc la plus grande fréquence de rotation) et les autres axes tels que

- les molécules linéaires (ou rotateurs linéaires)
- les toupies symétriques (ou rotateurs symétriques)
- les toupies sphériques (ou rotateurs sphériques)
- les toupies asymétriques (ou rotateurs asymétriques).
Molécules linéaires
Comme mentionné ci-dessus,
On pourra citer comme exemples de molécules linéaires : le dioxygène, le monoxyde de carbone, le radical hydroxyle (•OH), le dioxyde de carbone (CO2), le cyanure d'hydrogène, le sulfure de carbonyle (O=C=S), le chloréthyne (HC≡CCl) ou l'acétylène (HC≡CH).
Toupies symétriques
Une toupie symétrique est une molécule pour laquelle deux des moments d'inertie sont identiques. Pour des raisons historiques, les spectroscopistes divisent ces molécules en deux classes, les toupies symétriques oblates (en forme de disque) avec IA = IB < IC et les toupies symétriques prolates (en forme de cigare) avec IA < IB = IC. Leurs spectres sont assez différents, et sont immédiatement reconnaissables. Comme pour les molécules linéaires, la structure des toupies symétriques (longueurs de liaison et angles) peuvent être déduites de leurs spectres.
On peut citer comme exemples de toupies symétriques :
- oblates : le benzène (C6H6), le cyclobutadiène (C4H4), ou l'ammoniac (NH3).
- prolates : le chloroforme (CHCl3), le propyne (CH3C≡CH)
Toupies sphériques
Les toupies sphériques peuvent être considérées comme un cas spécial de toupies symétriques dont les trois moments d'inertie sont égaux (IA = IB = IC).
On peut citer comme exemples : le tétramère de phosphore P4, le tétrachlorure de carbone (CCl4), l'ion ammonium (NH4+) ou l'hexafluorure de soufre (SF6).
Toupies asymétriques
Une toupie asymétrique possède trois moments d'inertie différents. La plupart des grosses molécules sont des toupies asymétriques, même si elles présentent un fort degré de symétrie. Généralement, pour de telles molécules, une interprétation simple du spectre est impossible. Parfois, les toupies asymétriques présentent des spectres similaires à ceux d'une molécule linéaire ou d'une toupie symétrique, auquel cas la structure moléculaire doit aussi être similaire à celle d'une molécule linéaire ou d'une toupie symétrique. Pour le cas le plus général, cependant, tout ce qui peut être fait est de lisser le spectre sur trois moments d'inertie. Si la formule moléculaire est connue, on peut présupposer la structure probable, et à partir de cette structure, calculer les moments d'inertie. Si ceux-ci correspondent bien aux moments mesurés, on peut dire que la structure est déterminée. Dans cette approche, la substitution isotopique est invalide.
Quelques exemples : l'anthracène (C14H10), l'eau (H2O) - à cause des doublets non liants de l'oxygène central - ou le dioxyde d'azote (NO2) (pour des raisons similaires à l'eau).
Structure d'un spectre de rotation
Molécules linéaires
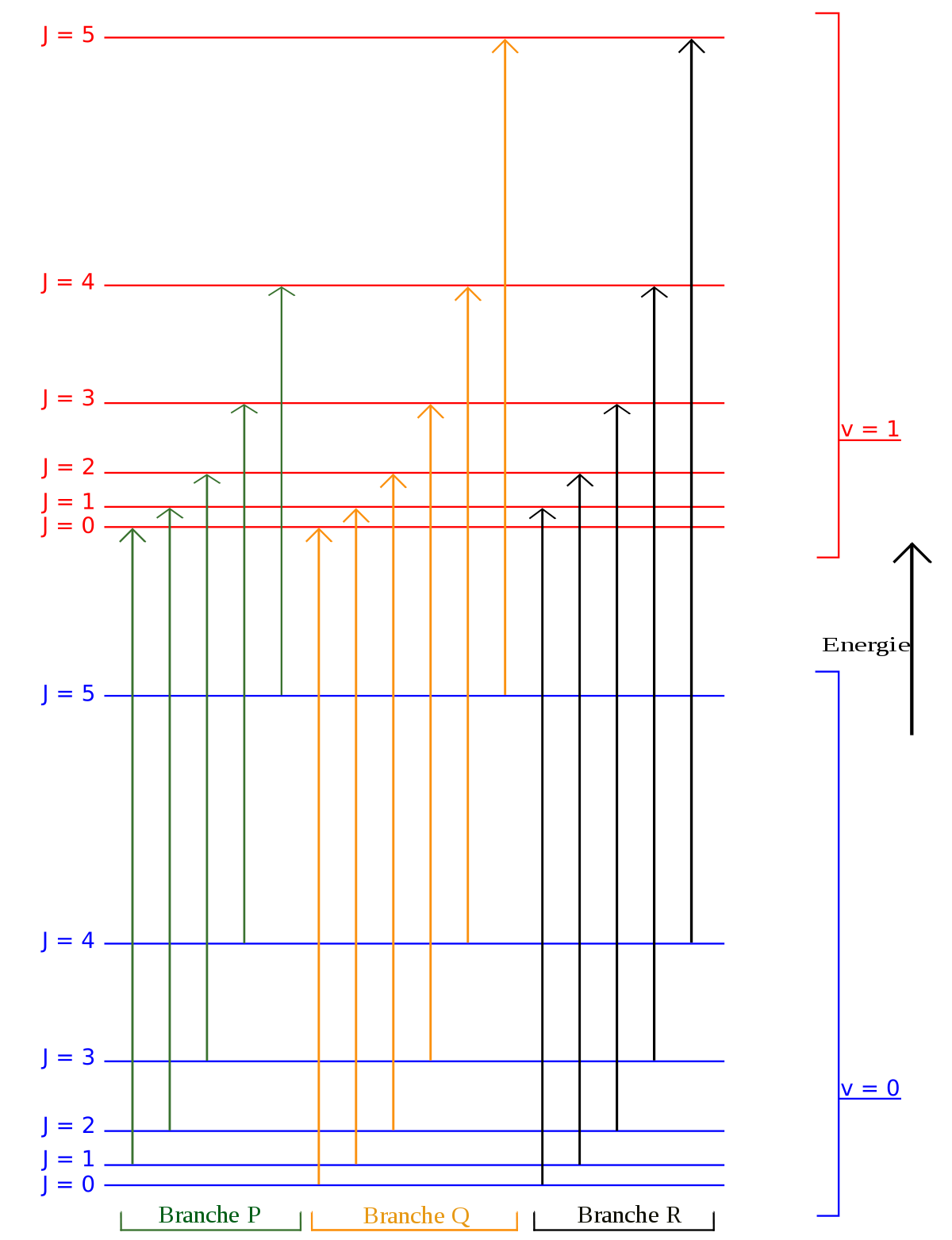
Ces molécules ont deux modes de rotation dégénérés (IB = IC, IA = 0). Comme on ne peut distinguer les deux modes, un seul nombre quantique de rotation (J) afin de décrire le mouvement de rotation d'une molécule.
Les niveaux d'énergie de rotation (


où


Les règles de sélection imposent que lors de la durée d'émission ou d'absorption le nombre quantique de rotation varie d'une unité, i.e.


où


On observe que, pour un rotateur rigide, les raies de transition sont espacées de manière égale dans l'espace des nombres d'onde. Cependant, ce n'est pas toujours le cas, sauf pour le modèle du rotateur rigide. Pour un modèle non rigide, on doit considérer les modifications du moment d'inertie de la molécule. Deux des raisons principales pour cette prise en compte sont :
- la distorsion centrifuge : lorsqu'une molécule tourne, la force centrifuge peut agir sur les atomes en les éloignant vers l'extérieur. Le moment d'inertie de la molécule augmente, donc
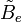
-
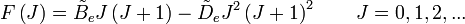
- où
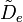
- L'espacement des raies pour le mode rotationnel est modifié en conséquence,
-
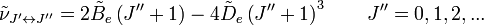
-
- l'effet de la vibration de rotation : une molécule est toujours en vibration. La molécule vibrant, ses moments d'inertie sont modifiés. Il existe de plus une force fictive, un couplage de Coriolis, entre le mouvement vibrationnel du noyau dans le repère en rotation (non-galiléen). Cependant, tant que le nombre quantique de vibration ne change pas (i.e. la molécule est dans un seul état de vibration), l'effet de la vibration sur la rotation n'est pas important, le temps de vibration étant beaucoup plu court que le temps de rotation. Le couplage de Coriolis est parfois négligeable également si l'on s'intéresse aux nombres quantiques de vibration et de rotation faibles.



Toupie symétrique
Le mouvement de rotation d'une molécule-toupie symétrique peut être décrit pas deux nombres quantiques de rotation indépendants (deux axes de rotation ayant les mêmes moments d'inertie, la rotation autour de ces axes ne nécessite qu'un nombre quantique de rotation pour une description complète). Au lieu de définir les deux nombres quantiques de rotation pour les deux axes indépendants, on associe l'un des nombres quantiques (J) avec le moment angulaire total de la molécule et l'autre (K) avec le moment angulaire de l'axe ayant un moment d'inertie différent des autres (i.e. l'axe C pour une toupie symétrique oblate et A pour une prolate). L'énergie de rotation


où



Les règles de sélection pour ces molécules fournissent des indications pour les transitions possibles, par :
-
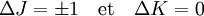

Cela est dû au fait que K est associé à l'axe de symétrie de la molécule et donc qu'elle ne possède pas de dipôle franc dans cette direction. Il n'y a donc pas d'interaction de ce mode avec les photons.
Cela donne les nombres d'ondes de transition :

ce qui est identique au cas d'une molécule linéaire.
Dans le cas des rotateurs non rigides, la correction de distorsion centrifuge au premier ordre est donnée par :

Les indices de la constante de distorsion centrifuge D indiquent que le mode de rotation impliqué et ne sont pas fonction du nombre quantique de rotation. La localisation des raies de transition sur un spectre est donnée par :

Toupie sphérique
Les molécules sphériques n'ayant pas de moment dipolaire, elles ne présentent pas de spectres purement rotationnels.
Toupie asymétrique
Le spectre de ce molécules présente habituellement de nombreuses raies en raison des trois modes de rotation différents et de leurs combinaisons. L'analyse suivante est valide pour le cas général et se réduit aux cas spéciaux précédents dans les limites appropriées.
A partir des moments d'inertie, on peut définir un paramètre asymétrique κ tel que :

variant de -1 pour une toupie symétrique prolate à 1 pour une toupie symétrique oblate.
On peut définir un hamiltonien de rotation réduit dépendant de J et κ. La représentation matricielle (symétrique) est quasi-diagonale, avec des coefficients nuls partout sauf sur la diagonale principale et la seconde sous-diagonale. Le hamiltonien peut être formulé de six manières différentes, selon l'orientation et l'arrangement des axes par rapport au référentiel absolu. Pour la plus asymétrique, la représentation des éléments diagonaux dans l'orientation main droite est, pour

- Hk,k(κ) = κk2
et les éléments non nuls hors diagonale principale (indépendants de κ) sot :
-
![H_{k,k+2}(\kappa)=\sqrt{[J(J+1)-(k+1)(k+2)][J(J+1)-k(k+1)]}/2](https://static.techno-science.net/illustration/Definitions/autres/7/784f25a8345f141702aceb4f625ba35d_f0eda81f6303568119cb2f092edd1ff2.png)
![H_{k,k+2}(\kappa)=\sqrt{[J(J+1)-(k+1)(k+2)][J(J+1)-k(k+1)]}/2](https://static.techno-science.net/illustrationWebp/Definitions/autres/7/784f25a8345f141702aceb4f625ba35d_f0eda81f6303568119cb2f092edd1ff2.png)
La diagonalisation de H donne un ensemble de 2J + 1 niveaux d'énergie rotationnelle Ek(κ) réduits. Les niveaux d'énergie rotationnelle pour le rotateur asymétrique de moment angulaire total J est alors donné par :

Interactions hyperfines
En plus de la structure principale observée dans les spectres microondes imputable au mouvement de rotation des molécules, un ensemble d'interactions plus « subtiles » sont responsables de petits détails dans ces spectres, et leurs études donne une compréhension vraiment profonde de la mécanique quantique des molécules. Les interactions principales responsables de ces légères modifications dans les spectres (séparations supplémentaires et déplacements de raies) sont dues aux interactions magnétiques et électrostatiques dans la molécule. Les forces particulières de ces interactions dans différentes molécules, mais en général, l'ordre de ces effets est (dans l'ordre décroissant) :
- interaction entre spins électroniques : se produit dans des molécules avec deux électrons non appariés ou plus, et est une interaction entre dipôles magnétiques.
- interaction entre spin électronique et rotation moléculaire : la rotation d'une molécule correspond à une dipôle magnétique qui interagit avec le moment magnétique dipolaire de l'électron.
- interaction entre spin électronique et spin nucléaire : il s'agit de l'interaction entre le moment magnétique dipolaire de l'électron et le moment magnétique dipolaire du noyau s'il existe.
- interaction entre gradient de champ électrique et quadrupôle électrique nucléaire, ou interaction entre le gradient de champ électrique du nuage électronique d'une molécule et les moments électriques quadrupolaires du noyau s'ils existent.
- interaction entre spins nucléaires : les moments magnétiques nucléaires interagissent entre eux.
Ces interactions donnent lieu à des niveaux d'énergies caractéristiques qui sont montrés par des techniques spectroscopiques de résonance magnétique comme la RMN ou la RPE, où ils représentent les « séparations à champ nul » qui sont toujours présentes.

















































